利用序列和组合图卷积网络预测蛋白质功能
2023-12-13秦琪琪丁学明王金雷
秦琪琪,丁学明,王金雷
(上海理工大学 光电信息与计算机工程学院,上海 200093)
1 引 言
蛋白质是生命的重要组成部分,具有不同功能的蛋白质在不同的生命活动中发挥作用,一旦缺失了关键蛋白质,生命过程将无法继续进行,并且蛋白质功能的准确预测可以帮助人类针对性的开发有效药物,因而了解蛋白质的功能对推进医学发展至关重要.蛋白质序列和结构隐含了其功能信息,高通量测序技术的快速发展加快了蛋白质序列数据的增长速度,但是蛋白质序列获得功能注释的速度远低于序列数据增长的速度,造成了已知蛋白质序列数量与已具有功能注释的蛋白质数量之间的差距不断增大,并且新序列缺乏实验验证的结构等生物信息.UniProt[1]数据库中已有2亿多条序列,而其中只有约为1%的序列是经过实验验证的[2],再次表明实验验证的速度远低于序列数据增长速度,无法通过实验验证快速注释蛋白质功能,因此通过计算方法来预测蛋白质功能开始成为探索蛋白质的重要步骤.传统的蛋白质功能预测方法不仅耗时耗力而且成本较高,无法达到较高的预测准确率,蛋白质序列数据的快速增长,深度学习的不断发展,都为准确预测蛋白质功能创造了条件,大量数据的学习和有效模型的构建可以大大提高预测准确率.
早期利用序列相似的方法预测蛋白质功能,比如BLAST[3]通过序列比对进行功能注释,然而高度不相似的蛋白质序列也可能具有相同的功能,所以这种方法预测准确率较低.针对该问题人们开始对BLAST方法进行改进,GOtcha[4]利用BLAST搜索结果进行功能注释,PFP[5]和ESG[6]使用E-value加权预测和机器学习的方法优化预测性能,都在BLAST的基础上提高了预测准确率,但是由于这些方法依然是依赖于序列相似性的,所以可靠性较低.因此人们开始结合蛋白质多种生物信息预测其功能,CombFunc[7]将ConFunc与蛋白质序列、结构域或蛋白质-蛋白质相互作用等信息结合进行功能预测,使用SVM组合这些信息,多种信息的结合使其预测性能优于BLAST,但仍然不能实现准确预测功能.随着数据的增长,机器学习算法和神经网络等方法被大量用于蛋白质功能预测.根据相互作用蛋白质共享功能的原理,蛋白质相互作用网络(PPI)[8-10]得到了人们的关注.Hishigaki[11]等人根据蛋白质相互作用图,引用n邻近蛋白质的方法,通过统计最大卡方值来预测蛋白质功能.2020年Cai Y等人提出了集成深度学习模型SDN2GO[12],该模型利用蛋白质序列、结构域和PPI 网络构建神经网络子模型,并通过加权分类器整合子模型进行功能预测.这些方法都使用了蛋白质结构和PPI网络等蛋白质序列之外的生物信息,而新测序的序列往往缺失这些丰富的生物信息,所以这些方法会受到相关信息缺失的限制.
由于蛋白质可具有多个功能,所以蛋白质功能预测可视为是一个多标签分类问题,适用于多标签分类的代表技术有支持向量机、决策树、K邻近方法和人工神经网络等[13],DeepGO[14]采用卷积神经网络(CNN)和PPI率先实现了较高的功能预测准确率,表明CNN可以从蛋白质序列和结构中提取有用信息,而图卷积神经网络(GCN)可以较好的解决CNN只能处理欧式数据的问题,结合新测序序列缺失丰富的生物信息问题,本文提出了基于蛋白质序列和组合图卷积网络的蛋白质功能预测模型(Protein Function Prediction using Sequences and Combined Graph Convolutional Networks,PFP-SCGCN),对GO ontology(GO)和Enzyme Commission(EC)两种蛋白质功能大类进行预测,并通过Grad-CAM(Gradient-weighted Class Activation Mapping)[15]方法分析PFP-SCGCN中的组合图卷积网络层识别功能位点.
2 方法概述
本文所提的蛋白质功能预测模型PFP-SCGCN主要由序列特征提取模块、进化信息提取模块、组合图卷积和全连接层块(FC blocks)组成,具体结构见图1,为了识别功能位点,利用Grad-CAM方法对组合图卷积的输出进行分析计算.
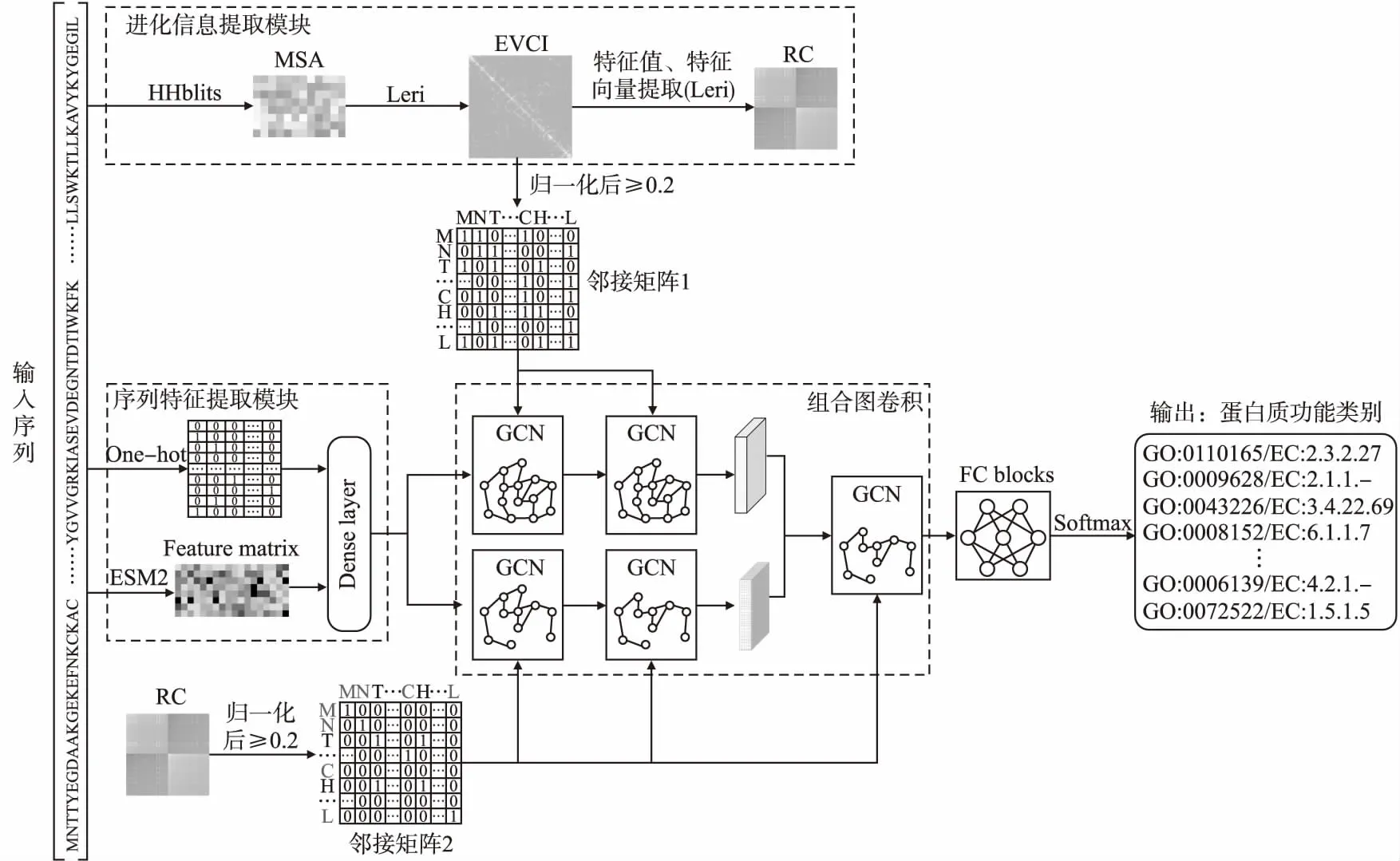
图1 PFP-SCGCN模型结构Fig.1 PFP-SCGCN model structure
2.1 序列特征提取
对于蛋白质序列,可以通过神经网络将序列氨基酸映射成空间向量,以此来获得丰富的信息.在PFP-SCGCN中先对蛋白质序列进行one-hot编码,在one-hot时考虑了20种常见氨基酸和空位gap,所以经过one-hot编码后的序列为S(L×21).由于考虑到当蛋白质序列较长时,其one-hot后的矩阵将会非常稀疏,不利于网络提取信息,所以将one-hot后的序列通过全连接层(Dense layer)进行信息稠密化,以此获得序列的空间嵌入信息.为获取更多的蛋白质序列信息,通过预训练好的蛋白质语言模型ESM2[16]将蛋白质序列中的每个氨基酸映射成空间表达式,L个氨基酸组成的序列经过ESM2处理后变为L个空间向量组成的特征矩阵(Feature matrix),维度为L×1280,该矩阵表达了序列氨基酸之间关联的隐层特征,将该隐层特征利用全连接层(Dense layer)进行再次映射,与获得的序列空间嵌入信息一起作为序列特征信息,即组合图卷积的初始输入.
2.2 进化信息提取
长期以来人们一直认为蛋白质的同源序列可以为了解其功能和结构提供重要的信息,比如在进化过程中保守的氨基酸通常与功能密切相关,所以同源序列隐含的进化信息可以为了解和设计蛋白质功能提供帮助,而且通过同源序列提取的进化信息有助于蛋白质的结构预测[17].除了通过进化信息探索蛋白质功能和结构,Ji-Yong An等人[18]利用进化信息和循环神经网络(RNN)预测自相互作用蛋白质,Shanwen Sun[19]等人利用进化信息,通过建立支持向量机分类器来识别抗冻蛋白,因此进化信息蕴含的丰富信息可以帮助人们更好地了解蛋白质.
目前已有较多的方法来提取蛋白质的进化信息,统计耦合分析(SCA)[20]通过对MSA的分析来识别共同进化的氨基酸对,为设计蛋白质折叠和识别功能区域提供了有用的信息.互信息(Mutual Information,MI)[21]可以识别蛋白质三维结构中一些直接相互作用的残基对,但是MI在计算耦合分数时只考虑了两个对齐列的位置.直接耦合分析(DCA)也可以在大量序列中寻找成对氨基酸之间的耦合关系[22].本文通过Leri[23]来提取进化信息,先为每条蛋白质序列搜索对应的多序列比对(MSA),Leri将MSA作为输入,考虑了MSA中所有位置氨基酸间的相互作用,通过序列产生器建模获得序列中任意氨基酸对之间的耦合项eij,序列中所有氨基酸对耦合项eij的组合形成进化耦合信息矩阵(Evolutionary coupling information,EVCI),对EVCI进行特征值计算,推断出与蛋白质功能相关的氨基酸残基群落(Residue Communities,RC).
2.2.1 多序列比对(MSA)
为了获取进化信息,利用多序列比对(MSA)来寻找同源序列,对于数据集中的序列,通过HHblits[24]工具在默认参数E-value为0.001的情况下,从Uniclust30[25]数据库中5次迭代搜索得到每一条蛋白质序列对应的MSA.
2.2.2 进化耦合信息EVCI提取
进化耦合信息(EVCI)包含了蛋白质序列中的所有氨基酸,EVCI中任意氨基酸对之间的耦合项eij,表达了进化过程中任意氨基酸对之间的相关程度,可以帮助人们判断每个氨基酸对于蛋白质功能或结构的贡献大小.获得每一条蛋白质序列的MSA后,利用Leri[23]提取对应的EVCI,具体内容如下:
1)通过HHblits工具得到每一条序列的MSA后,需要对MSA进行过滤处理,以去除MSA中低质量位点和序列.处理标准为两个,分别是当在MSA中的单个位点超过90%是空位gap时,移除该位点;当在MSA中的某条序列超过80%是空位gap时,移除该条序列.在MSA中每条序列的权重ω(τ)是由序列一致性(sequence identity,I)计算得到的,I是通过序列τ与其它所有序列之间的汉明距离(Hamming distance)DH(τ,τj)衡量的,具体权重计算如式(1)所示:
ω(τ)=(∑jI[DH(τ,τj)<θ])-1
(1)
其中参数θ为默认值0.2.
2)将处理后的MSA作为输入,利用基于伪似然最大化方法的全局概率模型[26]来捕获序列中氨基酸对之间的进化耦合关系.对蛋白质家族进化过程进行序列产生器建模,根据其家族中所有序列的空间分布,以概率P(τ)产生序列τ,P(τ)的计算为式(2):
(2)
其中Z为配分函数,通过将家族中所有序列的玻尔兹曼因数相加来进行归一化.每条序列的玻尔兹曼因数的测量是由进化统计能量E(τ)(马尔可夫随机场或Potts模型)定义的,具体如式(3)所示:
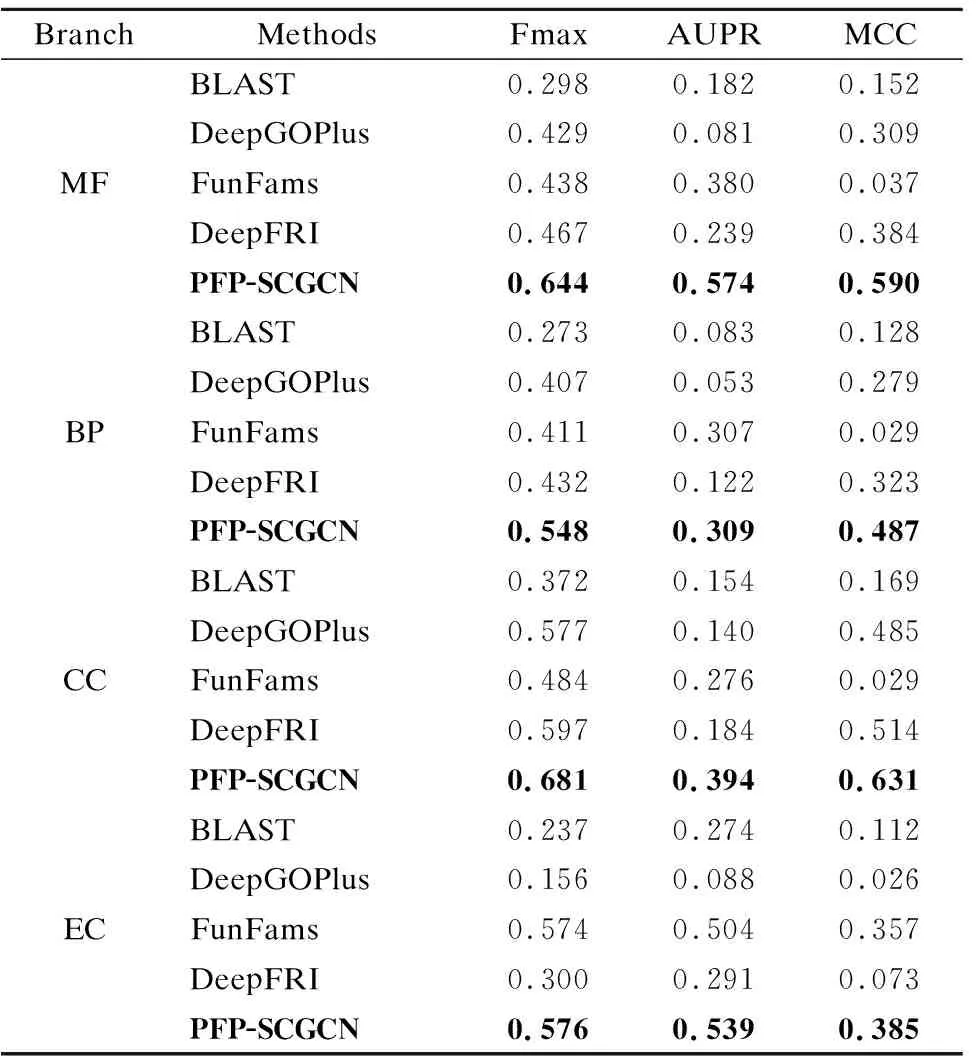
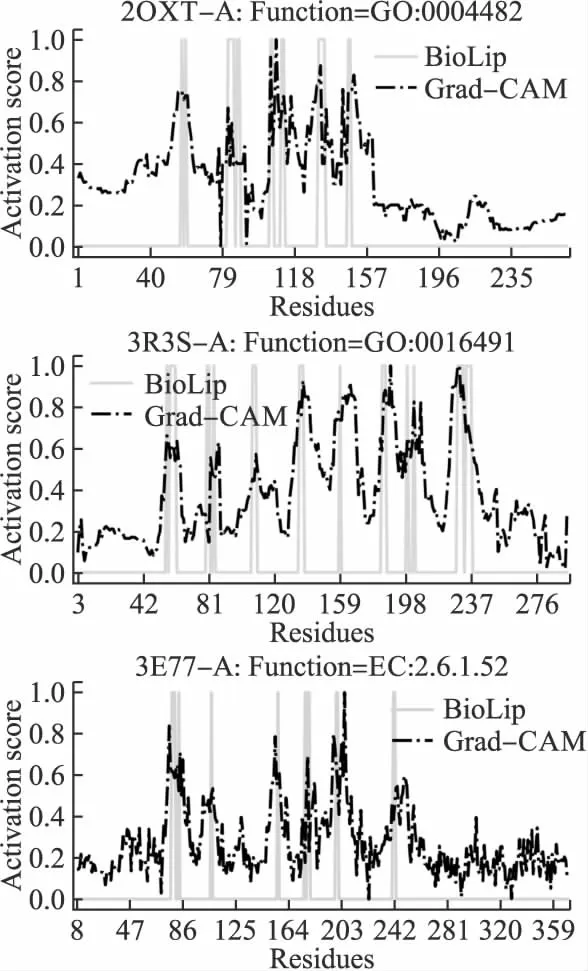
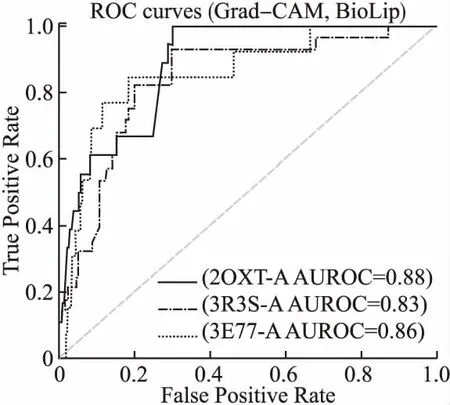
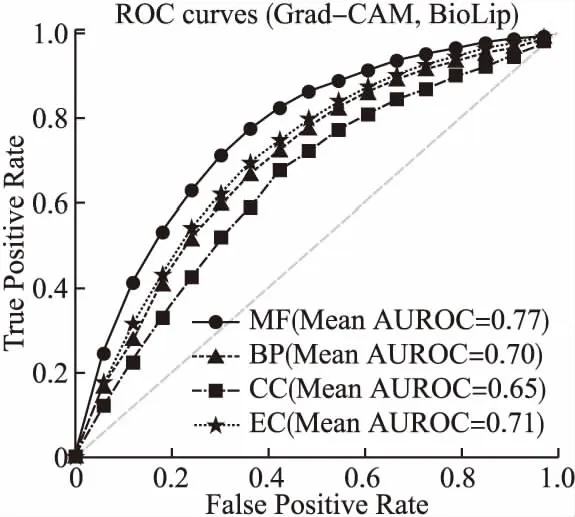
E(τ)=∑i (3) 其中eij为MSA中处于位置i的氨基酸与处于位置j的氨基酸之间的耦合项,hi为特定位点偏差项.eij和hi表达了两种类型的约束,分别是每对氨基酸之间的成对约束,以及特定位点偏差的约束,考虑了所有可能位置之间的相互作用,这是互信息没有考虑到的.对于eij和hi参数,利用位置分解的最大伪似然方法进行优化,如式(4)所示: (4) (5) 2.2.3 氨基酸残基群落RC提取 氨基酸残基群落(RC)包含的是蛋白质序列中的部分氨基酸,这部分氨基酸通常与蛋白质功能相关.RC用于表达进化过程中与功能相关的氨基酸残基对之间的耦合程度,因此可以利用RC信息着重关注序列中的部分氨基酸,去除部分噪声并减轻网络负担,使网络抓取与蛋白质功能相关的有用信息.Leri[23]利用谱分解法[27]从进化耦合信息(EVCI)中提取得到RC,谱分解法可以识别EVCI中的强耦合残基对,将氨基酸残基分类为不同的氨基酸残基群落(RC),具体内容如下: 对EVCI进行特征值计算和排序,通过雅可比迭代过程确定EVCI的正特征值和对应的特征向量,取其前5个特征值中的两个及其对应的特征向量来定义与功能相关的氨基酸残基群落(RC),将5个特征向量定义为Vk|k=1,2,…,5,本文使用的RC由两部分组成,分别定义为: 其中ε为默认参数0.05.图1中进化信息提取模块展示了使用Leri提取EVCI和RC的主要流程,其中MSA为N×L,代表搜索到的N条序列;EVCI为L×L,代表长度为L的序列中每对氨基酸之间的耦合关系;RC为m×m,是从EVCI中根据特征值计算得到的,代表了与功能相关的氨基酸残基之间的耦合关系,是蛋白质序列中的部分氨基酸且具有强耦合关系,所以m PFP-SCGCN模型采用组合图卷积网络分析蛋白质序列,组合图卷积网络定义为:采用3个通道的图卷积网络,先将其中两通道图卷积网络并行,并行的两通道图卷积分别由两层网络构成,每层网络输出的节点特征维度为512,进行并行的图卷积采用相同的初始输入,并且分别采用两种邻接矩阵来进行信息融合,实现信息特征的多样提取;接着使用另一通道图卷积网络与并行的两通道图卷积网络进行串联,用于处理并行图卷积的输出,进行串联的单通道图卷积由一层网络构成,该层网络输出的节点特征维度为512,初始输入为并行两通道图卷积的输出,邻接矩阵采用并行图卷积中使用的两种邻接矩阵中的一个,以此对并行图卷积输出的信息特征进行噪声过滤和蛋白质功能相关信息再次提取,加大了蛋白质序列中可能与功能相关的氨基酸节点的关注度,使进行串联的单通道图卷积输出包含丰富的功能相关的信息特征.3个通道图卷积的使用实现了图卷积网络的串并联组合,从而创建组合图卷积网络. 图卷积网络(GCN)在卷积神经网络(CNN)的基础上考虑了图结构信息[28],随着图数据的不断丰富,GCN已经被广泛应用于各个领域.图G(V,E,A)中,V代表了图中的节点集合,E表达了节点之间的连接边,A称为邻接矩阵,表达了节点之间是否存在连接.GCN通过层与层之间的传递来更新节点特征,如式(6)所示: fl+1=σ(fl,A) (6) 其中fl为GCN中第l层网络的输出,当l为0时,f0为网络的初始特征输入,每一层中的具体处理方式如式(7)所示: (7) 本文将蛋白质序列氨基酸视为图中节点,将提取的EVCI和RC在同等级别上进行归一化,值大于等于0.2阈值的对应位置视为氨基酸对之间存在联系,以此生成两种邻接矩阵,即邻接矩阵1和邻接矩阵2.EVCI衡量了蛋白质序列中所有氨基酸对之间的耦合关系,其包含了非常丰富的信息,RC衡量了蛋白质序列中部分氨基酸残基对之间的强耦合关系,其通常与蛋白质功能相关,所以RC去除了EVCI中的部分噪声.组合图卷积中并行的两通道GCN将序列特征提取模块提取的蛋白质序列特征信息作为初始输入f0,其中一个通道GCN采用了根据EVCI生成的邻接矩阵1,考虑序列中所有氨基酸之间的相互作用,而另一通道GCN采用根据RC生成的邻接矩阵2,只认为与功能相关的氨基酸之间存在联系,忽略其它氨基酸之间的作用.两通道GCN的并行将EVCI和RC的信息结合起来,实现了既通过RC去除EVCI中包含的部分噪声,又考虑EVCI中包含的可能有用的丰富信息,其中并行两通道GCN分别由两层512通道数的网络构成.并行GCN之后又采用一层通道数为512的GCN进行串联,串联GCN将并行GCN的输出作为输入,使用根据RC生成的邻接矩阵2,实现了噪声的再次过滤.组合图卷积的输出经过一个由3层全连接网络构成的分类器(FC blocks)进行分析,提取出蛋白质功能类别的分类特征,每层的通道数为1024,最后经过Softmax激活函数输出每个功能类别的预测概率. 设计电压比较电路,供5V直流电,用于比较的电路电阻R34.5kΩ、R41kΩ,可变电阻 R5初值 0.5kΩ,25℃时热敏电阻R210kΩ,UA点电压2.5V,UB点电压约3.75V,电压比较器LM311正极电压低于负极,不通,无输出。温度升高到60℃时,热敏电阻R2降到3kΩ,电压约1.2V,UA点电压升到3.8V,高于UB点电压,电压比较器正极电压高于负极,接通,输出信号。可变电阻用于设置比较电压的阈值,可变电阻阻值越大,UB点电压越低,报警温度越低。 氨基酸残基群落(RC)包含了与功能相关的氨基酸残基信息,为了探讨RC信息的实用性,利用Grad-CAM[15]方法计算特定氨基酸残基为功能位点的概率.Grad-CAM是一个类判别定位技术,为基于CNN模型的预测提供了视觉解释,可以利用该方法计算出网络对每个氨基酸的关注度,从而得到特定氨基酸残基为功能位点的概率.采用Grad-CAM根据组合图卷积的输出进行梯度值计算,得到序列中每个氨基酸为功能位点的置信分数,分数越高则表示其为功能位点的概率越高,并将预测功能位点结果与BioLip[29]进行对比,BioLip数据库包含了蛋白质真实功能位点信息.利用组合图卷积网络最后一层GCN输出的特征,首先计算输出特征图H中每个通道对于特定功能类别的权重值,如式(8)所示: (8) (9) 其中Hk,c为网络针对功能类别c的输出特征图H的k通道值,通过ReLU激活函数保留与功能类别c相关的特征. 对于众多的蛋白质功能,可广泛分为GO ontology(GO)和Enzyme Commission(EC),其中GO 可划分为3大类:分子功能(Molecular Function,MF)、生物过程(Biological Process,BP)、细胞成分(Cellular Component,CC),EC功能注释通过EC编号表达,EC编号由4个圆点隔开的数字组成.本文对GO和EC两种功能大类进行预测,从SIFTS[30]数据库中下载蛋白质功能注释信息和蛋白质序列,该数据库提供了来自蛋白质数据库PDB[31]和UniProt知识库(UniProtKB)[1]的信息,结合了蛋白质结构和序列的信息,有利于在蛋白质结构和序列之间传递蛋白质功能注释.在从SIFTS下载的数据中,对功能类别和序列进行筛选.对于GO功能注释保留根据EXP(Inferred from experiment)、IDA(Inferred from direct assay)、IPI(Inferred from physical interaction)、IMP(Inferred from mutant phenotype)、IGI(Inferred from genetic interaction)、IEP(Inferred from expression pattern)、TAS(Traceable author statement)和IC(Inferred by curator)获得的注释,并且为了保证每个功能类别能有足够的蛋白质数据,当具有某个功能类别的蛋白质数量介于50~5000之间时,就把该功能类别添加到总的功能类别中,EC功能注释只保留了三级和四级的EC编码.对于所下载数据集中的蛋白质序列,只保留含有20种常见氨基酸的序列,利用MMseqs2聚类工具以95%的序列一致性去冗余,并且序列长度小于等于1024. 处理后的数据集中,GO总共有34501个蛋白质,EC总共有18336个蛋白质,GO中MF,BP和CC总的功能类别数分别为5013个,12248个,1688个,EC的总功能类别数为577个.一个蛋白质可以具有多个功能,具有GO功能的蛋白质可以同时具有属于MF,BP,CC这3大类别的功能,有的则可能只具有属于其中两大类或者一类的功能,对数据集随机打乱后按照大约8∶1∶1的比例划分数据集,具体的数据分布如表1所示. 表1 GO和EC的数据集分布Table 1 GO and EC data set distribution 精准率(Precision),召回率(Recall)和F1-score是常用的分类评价指标,Precision表达了对蛋白质预测出的功能类别中为真实功能类别的概率,Recall表达了蛋白质的真实功能类别中被正确预测出的个数占比,F1-score则根据Precision和Recall的值计算得出,计算方式为式(10): (10) F1-score综合评价了Precision和Recall. (11) 其中TP为正确预测为正样本的个数,TN为正确预测为负样本的个数,FP为错将负样本预测为正样本的个数,FN为错将正样本预测为负样本的个数. 本文基于TensorFlow2框架搭建PFP-SCGCN模型,通过NVIDIA GeForce RTX 2080Ti GPU进行模型训练,选择Adam优化器来优化模型参数,设置学习率为0.001,batch_size为64,训练周期为100 epoch,为了防止模型过拟合,采用early stopping机制,周期为10,损失函数为二进制交叉熵损失函数,计算方式如式(12)所示: (1-yij)log(1-P(yij))] (12) 其中N为总共的样本数,C指总共的功能类别数,yij为蛋白质i对于功能类别j的真实标签,P(yij)则为其对应的预测概率. 模型PFP-SCGCN主要与5个方法进行对比,包括BLAST[3]、DeepGOPlus[32]、FunFams[33]、DeepFRI[34]和GAT-GO[35],具体如下: 1)BLAST[3]:属于无监督方法,利用序列相似性进行功能注释,本文采用了与CAFA1[36]中相同的BLAST使用方法.设E-value为默认值1e-3,将训练集中序列的功能注释给对应的测试集序列,预测分数为序列一致性分数. 2)DeepGOPlus[32]:使用蛋白质序列信息进行功能预测,为基于一维卷积神经网络(1DCNN)的模型,将通过one-hot编码后的序列经过16层不同卷积核大小的1DCNN进行蛋白质功能预测. 3)FunFams[33]:属于无监督方法,是一种基于域的蛋白质功能注释方法,利用CATH superfamilies的功能分类进行蛋白质功能注释,对每个蛋白质序列通过HMMER3[37]工具在CATH FunFams中搜索,将HMM得分最高的功能类别注释给测试序列,计算搜索结果中功能类别出现的频率为预测分数,并且GO功能类别预测分数按照Das[33]等人所述在GO层次结构中向上传播. 4)DeepFRI[34]:使用蛋白质序列信息和结构信息作为模型的输入,通过预训练好的 LSTM(Long Short Term Memory)模型提取序列特征,根据蛋白质结构构建氨基酸之间的接触图,将LSTM的输出与构建的接触图一起输入给GCN来预测蛋白质功能. 5)GAT-GO[35]:使用蛋白质序列信息和预测的残基接触图作为模型的输入.首先生成每条蛋白质序列对应的PSSM(Position Specific Scoring Matrix),再根据PSSM通过RaptorX[38]预测对应的残基接触图,对蛋白质序列进行one-hot编码,并利用ESM-1b[16]获得残基级嵌入信息和蛋白质嵌入信息,通过一维卷积神经网络(1DCNN)处理序列one-hot编码、PSSM和残基级嵌入信息,将1DCNN的输出和预测的残基接触图通过图注意力网络(GAT)进行信息融合,最后将GAT的输出与蛋白质嵌入信息输入给全连接层进行蛋白质功能预测. 根据Fmax,AUPR和MCC值的比较,对比PFP-SCGCN与BLAST、DeepGOPlus、FunFams、DeepFRI和GAT-GO这5种方法的预测性能差异,所对比方法均按照其默认参数进行设置,不同方法在MF,BP,CC和EC中的预测性能对比如表2,结果表明模型PFP-SCGCN在所有评价指标中几乎都达到了最好的性能.对比结果中DeepFRI各项指标值排名靠前,这与其使用了蛋白质结构信息有关,PFP-SCGCN的各项指标值远高于DeepFRI,表明了PFP-SCGCN只用序列信息而不用结构等其它生物信息就可以较准确的预测功能.这是因为DeepFRI中使用的残基接触图是蛋白质三级结构到二维空间的映射,在残基接触图中存在连接的氨基酸对只能代表其在空间中距离较近,但其不一定是功能结合位点,而PFP-SCGCN中使用了进化耦合信息(EVCI)和氨基酸残基群落(RC),其中EVCI包含了非常丰富的信息,衡量了蛋白质序列在进化过程中氨基酸对之间的关联程度,进化过程中保守的氨基酸很有可能与蛋白质功能相关,EVCI中较大的值对应位置的氨基酸对具有强耦合关系,说明该氨基酸对在进化过程中关联密切,则其可能与蛋白质功能有很大的相关性,RC包含了EVCI中具有强耦合关系的氨基酸,故RC拥有蛋白质功能相关信息,过滤了EVCI中的部分噪声.PFP-SCGCN在组合图卷积网络中同时使用了EVCI和RC,既考虑了EVCI中的多样信息,又通过RC引导网络关注与功能相关的氨基酸节点,以此使网络极大程度的开发与蛋白质功能相关的重要信息,从而使PFP-SCGCN更好的预测蛋白质功能.虽然GAT-GO模型输入中采用了预测的残基接触图,但是在GO中GAT-GO的预测性能整体优于DeepFRI,这与其使用了GAT网络有关,GAT中的自注意力机制给图中每个节点分配不同程度的注意力,减少了对图结构的依赖程度,因而使GAT-GO在使用预测的残基接触图时也可以较准确的预测蛋白质功能,但是预测的残基接触图难免含有噪声,并且预测出具有接触关系的残基对不一定是与功能相关的.FunFams在EC中的预测准确率排名第二,这与其使用的CATH superfamilies数据有关,使EC功能类别出现的频率较高.BLAST方法的预测性能在这5个对比方法中较差,这也表明仅基于序列相似性预测功能是不够准确的.DeepGOPlus的性能高于BLAST,表明CNN可以从序列中提取有用的信息,但对于准确预测功能还有距离. 表2 不同方法的功能预测性能比较Table 2 Performance comparison of function prediction between different methods 为了检验PFP-SCGCN的鲁棒性,计算测试集中的序列与训练集中所有序列的序列一致性,按照序列一致性的最大值将GO和EC的测试集划分为了30%,40%,50%,70%,95%这5组,选择最大序列一致性≤30%这组测试集的预测性能进行比较,表3展示了具体结果.从表3对比结果可以看出,当测试集序列与训练集序列极度不相似时,所提模型PFP-SCGCN依然可以达到较高的预测准确率,并且整体的评价指标值都优于对比方法,证明了PFP-SCGCN具有较好的鲁棒性.PFP-SCGCN在≤30%序列一致性测试集的预测性能优于DeepFRI,再次证明了PFP-SCGCN不使用蛋白质结构这种序列信息之外的生物信息,依然可以较准确的预测蛋白质功能.BLAST的预测性能在鲁棒性对比结果中依然是较差的,表明了仅基于序列相似性进行功能预测的不可靠性.DeepGOPlus在鲁棒性检验中的性能较稳定,表明多层CNN结构是可以提取部分有价值信息的,但仍需改进. 表3 在≤30%序列一致性中的预测性能比较Table 3 Comparison of prediction performance in ≤30% sequence identity 图2 功能位点识别示例Fig.2 Examples of functional site identification 图3 功能位点识别示例对应ROC曲线Fig.3 ROC curves of the examples of functional site identification 图4 功能位点识别评估Fig.4 Functional site identification evaluation 序列提取技术的快速发展使蛋白质序列数据增长速度加快,导致未功能注释的蛋白质数量不断增多,并且新测序序列缺失丰富的生物信息,针对该问题,本文提出了仅利用蛋白质序列信息的蛋白质功能预测模型PFP-SCGCN.该模型利用one-hot编码,预训练好的蛋白质语言模型ESM2和全连接网络提取蛋白质序列特征信息,通过MSA提取进化耦合信息(EVCI)和氨基酸残基群落(RC),获得序列中氨基酸对之间的耦合关系,构建并行两通道GCN和串联单通道GCN结合的组合图卷积网络,根据EVCI和RC生成表达氨基酸之间连接关系的两种邻接矩阵,将这两种邻接矩阵与序列特征信息一起作为组合图卷积网络的输入,最后通过多层全连接网络和Softmax激活函数预测蛋白质功能类别,并通过Grad-CAM方法识别功能位点.PFP-SCGCN在MF,BP,CC和EC数据上的预测性能均优于对比方法,并且具有较好的鲁棒性,表明PFP-SCGCN可以在仅使用序列信息的情况下较准确的预测蛋白质功能.Grad-CAM计算值与BioLip的重合结果表明RC为PFP-SCGCN提供了有用的信息,使PFP-SCGCN具有一定的功能位点注释能力. 虽然PFP-SCGCN已具有较好的功能预测能力和功能位点注释能力,但EVCI和RC信息的使用仍有改进空间,并且PFP-SCGCN的功能位点识别能力可以进一步的提高,未来将尝试不同的信息提取方式和新的网络架构,以此进一步优化模型性能.PFP-SCGCN相关代码可在https://github.com/psxz1/PFP-SCGCN获得.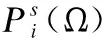
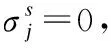
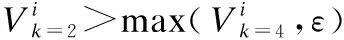
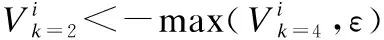
2.3 组合图卷积和全连接层块(FC blocks)
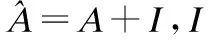
2.4 功能位点识别

3 结果分析
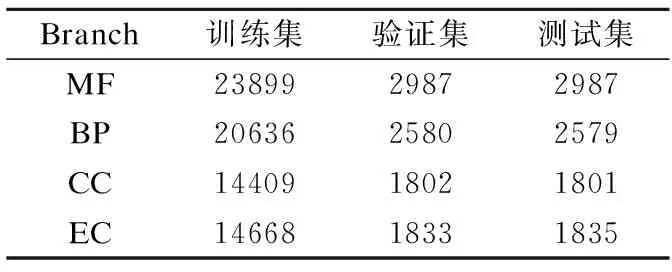
3.1 构建数据集
3.2 评价指标
3.3 实验环境及参数设置
3.4 对比实验
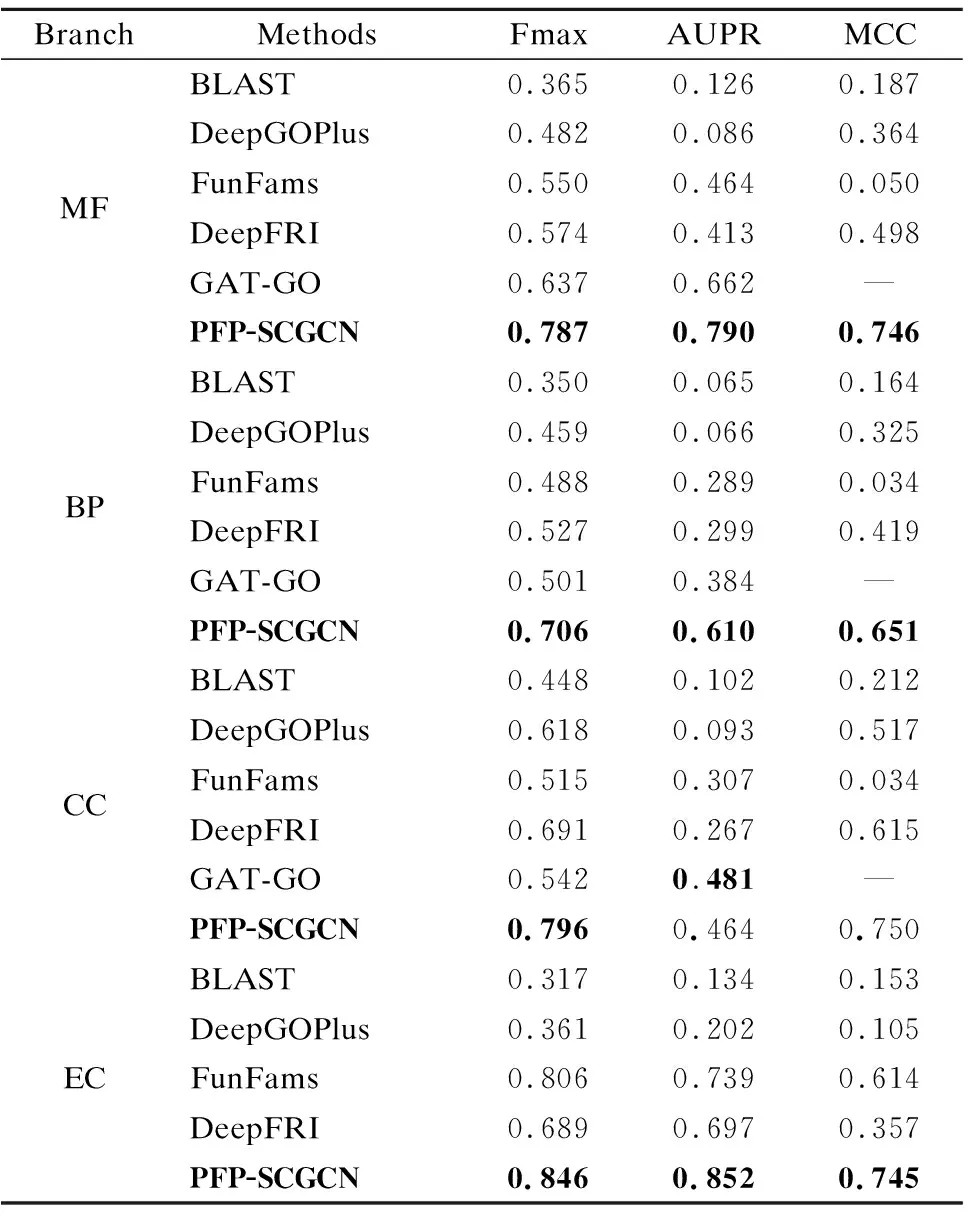
3.5 实验结果对比分析
3.6 模型鲁棒性检验
3.7 功能位点识别
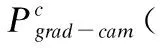
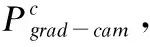
4 结束语
