铁死亡及其在炎症性肠病中对肠上皮细胞作用机制的研究进展
2023-12-12陈双兰刘青松胡双元
陈双兰,刘青松,胡双元,张 怡,刘 蓉
(成都中医药大学 1.附属医院消化内科、2.药学院,四川 成都 610072)
铁死亡是2012年由Dixon等[1]提出的一种铁依赖性、脂质氧化介导的程序性细胞死亡新形式,其典型特征是脂质过氧化产物的积累和膜多不饱和脂肪酸(polyunsaturated fatty acid,PUFA)的消耗。炎症性肠病(inflammatory bowel disease,IBD)是一组原因不明的非特异性慢性肠道炎性疾病,包括溃疡性结肠炎(ulcerative colitis,UC)、克罗恩病(Crohn’s disease,CD)和未定型结肠炎。UC是累及结肠黏膜层和黏膜下层的连续性炎症,而CD是可累及整个消化道全层的非连续性炎症,最常累及部位为末端回肠、结肠和肛周。IBD发病机制目前倾向于人为环境触发因素,主要是肠道菌群的异常在遗传易感宿主中引起的黏膜免疫系统不受控制的激活而诱发了慢性肠道炎症。主要临床表现是腹泻、腹痛,甚至可有脓血便。
研究发现,大量的铁摄入会增加患UC的风险并加重UC患者的临床症状,且UC黏膜中活性氧(reactive oxygen species,ROS)的生成与疾病活动成正比,而铁螯合剂是一种铁死亡抑制剂可以减少ROS的产生并改善肠道炎症。这些提示IBD可能与铁死亡之间存在联系:肠内过量的铁积累通过芬顿(Fenton)反应产生ROS,引起氧化应激,而脂质过氧化的程序性出现使得肠上皮细胞(intestinal epithelial cells,IEC)发生铁死亡,破坏肠黏膜机械屏障,导致IBD的发生。进一步地,在UC中观察到升高的铁蛋白重链(ferritin heavy chain,FTH)信号主要集中在IEC,这提示铁死亡主要发生在IEC中,且通过透射电镜能观察到IEC的线粒体萎缩,这与铁死亡形态学特征一致。
就肠道而言,IEC死亡破坏肠道完整性,使肠道物理屏障被氧化应激损伤,随着细胞与周围环境之间的相互作用,肠道化学屏障、免疫屏障和生物屏障被破坏,进而出现一系列的肠功能障碍。因此,阐明铁死亡机制及其对IEC的作用就显得尤为重要。
1 铁死亡及其机制
1.1 铁死亡概述铁死亡是不同于细胞凋亡和坏死的细胞死亡新形式。它的特征是[2]:①形态学上,线粒体萎缩、线粒体膜密度增加、线粒体嵴减少或消失;②生物化学上,铁积累、ROS的产生和过度的脂质过氧化;③涉及一组特殊的基因参与,如前列腺素-内过氧化物合酶2 (prostaglandin-endoperoxidase synthase 2,PTGS2)、长链脂酰辅酶A合成酶家族成员4(acyl-CoA synthetase long chain family member 4,ACSL4)等。铁死亡诱导剂目前可分为四类[3]:①谷胱甘肽(glutathione,GSH)清除剂,Erastin通过抑制胱氨酸-谷氨酸反向转运体(cystine/glutamate antiporter,system Xc-)来阻止胱氨酸摄取,从而消耗GSH;②谷胱甘肽过氧化物酶4 (glutathione peroxidase 4,GPX4)抑制剂,这类诱导剂包括RSL3、Altretamine 和DPI17,均可以直接抑制GPX4的活性;③FIN56,一方面FIN56以尚不清楚的方式诱导GPX4的降解,另外一方面,FIN56激活鲨烯合成酶,进而消耗甲羟戊酸途径中的辅酶Q10 (coenzyme Q10,CoQ10),削弱细胞抗氧化能力,触发铁死亡;④FINO2,是一种可以优先氧化胞内不稳定铁的亲脂性过氧化物,并进一步导致PUFAs的广泛氧化,此外,FINO2可以抑制GPX4,降低其活性和蛋白水平。关于铁死亡抑制剂[3]:①铁螯合剂,它与铁离子结合形成复合物,降低胞内不稳定的游离铁,从而减轻脂质过氧化;②β-巯基乙醇,通过与胱氨酸形成二硫化物促进系统Xc-的激活而限制了Erastin诱导的铁死亡;③捕获自由基的抗氧化剂,包括维生素E和基于芳香胺的ferrostatin-1(Fer-1)和liproxstatin-1(Lip-1),这类抗氧化剂能阻止自由基的级联传播,保护脂质免受自氧化;④脂氧合酶(lipoxygenase,LOX)抑制剂,可抵消LOXs催化的脂质过氧化作用。
1.2 铁死亡机制(Fig 1)。
1.2.1促铁死亡机制
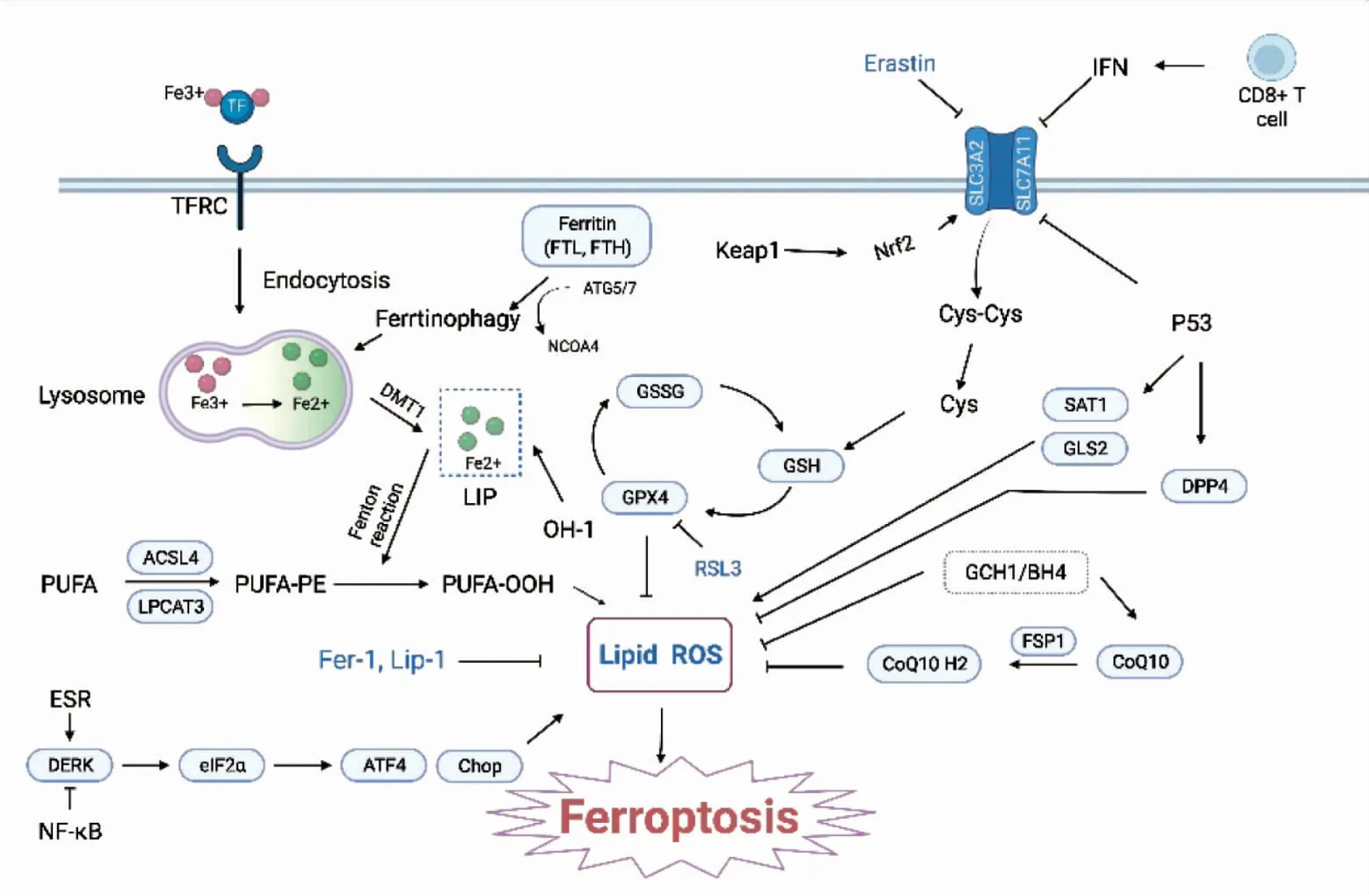
Fig 1 Mechanism of ferroptosis →represents promotion. ┫ represents inhibition.
1.2.1.1 铁蛋白与铁死亡 铁依赖的脂质过氧化物作用是铁死亡的标志信号之一。Fe3+通过转铁蛋白受体(transferrin receptor,TFRC)和内吞作用(Endocytosis)进入细胞,在还原成Fe2+后由二价金属转运体蛋白1(divalent metal transporter 1,DMT1)协助流入胞质的不稳定铁池(labile iron pool,LIP)。铁蛋白是铁的惰性储存形式,一般不能促进脂质过氧化,但铁超载过重时,铁蛋白水解为Fe2+,促进Fenton反应和ROS的生成,细胞内的Fe2+则储存在LIP中,导致对铁死亡的敏感性增加。另外,铁蛋白可通过靶向自噬,导致铁蛋白溶酶体(ferritin lysosome)降解,将铁释放到LIP中,从而增加铁死亡的敏感性。
1.2.1.2 ACSL4与铁死亡 ACSL4是ACSL家族的一员,在体内催化合成脂酰辅酶A,这是脂肪酸分解代谢的第一步。含多不饱和脂肪酸的磷脂(polyunsaturated fatty acid phospholipids,PUFA-PLs)可通过酶促和非酶促途径氧化。脂氧合酶依赖的酶促途径包括ACSL4和溶血磷脂酰胆碱酰基转移酶3(lysophosphatidyl cholinyltransferase 3,LPCAT3)的激活,而在铁死亡过程中,此途径对PUFA-PLs生成脂质过氧化物起着关键作用。Doll等[4]发现ACSL4将长链PUFAs活化以参与膜磷脂的合成,这些膜上的长链PUFAs容易被氧化,而ACSL4就是通过参与合成这些易被氧化的膜磷脂而成为引发铁死亡的必需组分之一,使得细胞对铁死亡激活剂RSL3等诱导因素敏感。在肿瘤细胞中,放疗可以产生大量的ROS,并上调ACSL4的表达,共同促进脂质过氧化,最终使肿瘤细胞发生铁死亡,而敲除ACSL4基因可导致明显的放射抵抗。
1.2.1.3 内质网应激与铁死亡 蛋白激酶样内质网激酶(protein kinase-like endoplasmic reticulum kinase,PERK)是内质网应激的主要传感器,在内质网应激反应中,PERK通过磷酸化被激活,进而激活并调控真核起始因子2α亚基(eukaryotic initiation factor 2α,eIF2α),并促进转录因子4(activating transcription factor 4,ATF4)和C/EBP同源蛋白(C/EBP-homologous ER protein,Chop)的产生。PERK-eIF2α信号通路通过调控ROS的产生在细胞铁死亡过程中发挥作用。Xu等[5]用RSL3处理细胞观察到铁死亡发生的同时发生了eIF2α的磷酸化、ATF4和Chop的上调,之后再将PERK的选择性抑制剂GSK 414添加到细胞中,发现peIF2α、ATF4和Chop的表达不仅受到抑制,还降低了RSL3刺激后细胞的铁死亡。综上研究表明了内质网应激在细胞铁死亡中的促进作用。另外有越来越多的证据表明核因子κB(nuclear factor kappa-B,NF-κB)通路可能参与了内质网应激信号转导和细胞铁死亡的调控。
1.2.1.4 自噬与铁死亡 最近也有研究将铁死亡定义为一种自噬依赖的细胞死亡。Hou等[6]证明自噬通过降解成纤维细胞和癌细胞中的铁蛋白促进铁死亡,通过敲除自噬相关基因Atg5和Atg7,降低了脂质过氧化及细胞内Fe2+水平,限制了Erastin诱导的细胞铁死亡,且Atg5介导的自噬是铁蛋白降解所必需的,核受体共激活剂4(nuclear receptor coactivator 4,NCOA4)是铁蛋白自噬转化的选择性转运受体,NCOA4的遗传抑制削弱了铁蛋白降解及随后的铁死亡。脂吞噬是一种选择性自噬,脂吞噬介导的细胞内脂滴自噬降解促进脂质过氧化,诱导细胞铁死亡[7],以上均为自噬和铁死亡之间的联系提供新证据。
1.2.1.5 免疫细胞与铁死亡 近年来,在癌症研究中发现CD8+T淋巴细胞通过诱导铁死亡和焦亡来抑制肿瘤。具体来讲,Wang等[8]发现,免疫治疗激活CD8+T淋巴细胞释放的干扰素γ (interferon-γ,IFNγ) 下调调节型溶质载体家族3成员2(Solute carrier family 3 member 2,SLC3A2)和催化型溶质载体家族7成员11(Solute carrier family 7 member 11,SLC7A11)的表达,限制了肿瘤细胞对胱氨酸的摄取,进而促进了肿瘤细胞的脂质过氧化及随后的铁死亡,这有助于抗肿瘤效果。
1.2.2抑铁死亡机制
1.2.2.1 GPX4-GSH通路与铁死亡 铁死亡是一种由磷脂过氧化引起的细胞死亡。GSH依赖的磷脂过氧化物酶—GPX4是首个被发现的铁死亡中枢抑制剂,是唯一能催化氧化生物脂还原的硒蛋白。GSH是一种含巯基的三肽,是GPX4不可缺少的辅助因子。GPX4利用GSH消除磷脂过氧化物来保护细胞免受铁死亡。GPX4是一种硒蛋白,而向动物或细胞输送硒可以抑制铁死亡,这提示硒可以影响铁死亡的敏感性。
1.2.2.2 FSP1-CoQ10通路与铁死亡 甲戊酸途径衍生CoQ10,不仅是线粒体电子传递链的关键部分,而且在线粒体外通过抑制自由基中间体来抑制脂质过氧化,因此CoQ10的激活能使铁死亡受到抑制。已经有文献报道在GPX4缺失的情况下,铁死亡抑制蛋白1(ferroptosis suppressor protein 1,FSP1)也可通过再生减少的CoQ10来阻断脂质过氧化并抑制铁死亡,这提供了一种新的独立的FSP1-CoQ10铁死亡途径[9]。
1.2.2.3 GCH1-BH4-磷脂通路与铁死亡 此通路由三磷酸鸟苷环水解酶-1(guanosine triphosphate cyclohydrolase 1,GCH1)及其代谢衍生物四氢生物蝶呤(tetrahydrobiopterin,BH4)组成。最近一篇文献报道,表达GCH1的细胞通过合成BH4引起脂质重塑,并选择性地防止PUFAs尾部的两个磷脂消耗来抑制铁死亡。Kraft等[10]通过脂质组学分析,GCH1表达的细胞还可在诱导后使减少的CoQ10再生;同样在他们的实验中,GCH1在GPX4敲除细胞中过表达并主要通过BH4的抗氧化作用来保护细胞免受铁死亡,也证明了这是一条完全独立于GPX4介导的铁死亡抑制途径。
1.2.2.4 Keap1-Nrf2通路与铁死亡 核转录因子E2相关因子2(nuclear transcription factor E2-related factor 2,Nrf2)是细胞抗氧化反应的关键转录因子,其靶点在铁代谢和脂代谢中发挥重要作用。Kelch样环氧氯丙烷相关蛋白 1(Kelch-1ike ECH-associated protein 1,Keap1)是Nrf2的特异性受体,经氧化应激后其构象发生变化,不与Nrf2相互作用,从而阻止其降解,积累的Nrf2转移到细胞核,并促进相关基因转录,如谷氨酸-半胱氨酸连接酶修饰亚基(glutamate-cysteine ligase modified subunit,GCLM)和GPXs,从而促进系统Xc-。Nrf2上调铁蛋白转录基因,增加铁储存和减少不稳定铁来调控铁稳态,也通过调节运铁素(ferroportin-1,FPN1)的表达来改变不稳定铁进出细胞,因此Nrf2的激活可以平衡细胞铁超载的情况,并防止氧化应激。激活的Nrf2还可上调GPXs改善细胞脂质过氧化,黄芩素被证明通过防止Nrf2降解和提高GPX4水平来缓解铁死亡。其它的,Nrf2通过诱导的铁相关靶基因血红素氧合酶1 (heme oxygenase-1,HO-1)、金属硫蛋白1G(metallothionein,MT-1G)和FTH1的表达对铁死亡具有保护作用。但较为矛盾的是,已有研究证明NRF2-HO-1通路的过度激活会干扰铁离子代谢导致铁死亡[11]。
1.2.2.5 NF-κB通路与铁死亡 NF-κB通路在调节细胞存活和增殖中发挥着关键作用。在人乳腺癌细胞中,核因子kappa B p65亚基( nuclear factor kappa B p65 subunit,NF-κB p65)的磷酸化抑制了PERK介导的铁死亡,而NF-κB信号通路的抑制导致了细胞的铁死亡,在IEC中也是如此[5,11]。Yao等[12]也发现,在肝癌细胞中,NF-κB的激活导致铁螯合因子LCN2的上调,LCN2通过消耗铁使铁死亡受到抑制。另外,铁死亡诱导剂Erastin可以通过抑制NF-κB通路来控制脓毒血症的发展[13]。然而,在胶质母细胞瘤细胞中,NF-κB激活却通过下调ATF4的表达促进铁死亡[14],这与之前的研究似乎是矛盾的,因此NF-κB通路的具体机制还需进一步阐明。
1.2.2.6 系统Xc-与铁死亡 系统Xc-由SLC7A11及SLC3A2组成[1]。细胞中的胱氨酸(cystinol,Cys-Cys)转化为半胱氨酸(cysteine,Cys),这对GSH的合成至关重要,而GSH可通过减少ROS及活性氮以降低细胞氧化损伤,避免铁死亡;但当谷氨酸浓度过高时,将会抑制系统Xc-,使Cys-Cys吸收不足,进一步使细胞中Cys不足,耗尽细胞中抗氧化的GSH,并导致依赖GSH的过氧化物酶GPX4失效,导致脂质过氧化物的积累并诱导铁死亡。除此之外,Erastin可通过与溶质载体家族7成员5(Solute carrier family 7 member 5,SLC7A5)结合抑制系统Xc-活性,发挥促铁死亡作用[1]。
1.2.3双向调节铁死亡机制
1.2.3.1 P53与铁死亡 P53是已经被广泛研究的肿瘤抑制基因,在所有恶性肿瘤中,50%以上会出现该基因的突变。由这种基因编码的蛋白质是一种转录因子,通过介导细胞周期阻滞、凋亡和衰老参与肿瘤的抑制,但近期越来越多的发现证明,P53还通过代谢活动参与肿瘤抑制。一方面,P53四个乙酰化位点的突变会完全破坏其调节代谢靶点的能力,如凋亡调节因子IGAR蛋白和SLC7A11,其机制是P533KR是乙酰化缺陷突变体,它不诱导细胞周期阻滞或凋亡但能下调SLC7A11促进谷胱甘肽耗尽,并在ROS诱导的应激下促进铁死亡;P53还可通过提高亚精胺/精胺N1乙酰转移酶1(spermidine/spermine N1-acetyltransferase 1,SAT1)和谷氨酰胺酶2(glutaminase,GLS2)的表达使细胞对铁死亡敏感;或者通过转录上调线粒体GLS2来促进铁死亡;ALOX12是位于人类染色体17P13.1非常接近于P53的基因,研究数据显示,P53通过SLC7A11的转录抑制间接激活ALOX12的功能,并导致ROS的积累和铁死亡[15]。另一方面,在大肠癌中,P53的缺失阻止了二肽基肽酶-4(dipeptidyl peptidase-4,DPP4)的核积累,从而促进质膜相关的DPP4依赖的脂质过氧化,并诱导铁死亡,这说明在大肠癌中,P53起负反馈调节;P53还通过介导肿瘤抑制因子周期蛋白依赖性激酶抑制剂1A(cyclin-dependent kinase inhibitor 1A,CDKN1A)的表达,延迟了因胱氨酸缺乏而引起的铁死亡[16]。综上,P53可能在铁死亡过程中起着双向调节作用。
1.2.3.2 HO-1与铁死亡 HO-1是一种催化血红素生成Fe2+、胆绿素和一氧化碳的Ⅱ相酶。研究证明HO-1具有保护细胞、抗炎、抗氧化、抗凋亡、抗增殖的作用。Adedoyin等[17]发现在肾小管细胞中HO-1的过表达减轻了铁死亡,而敲除HO-1可以增强Erastin诱导的铁死亡。但相反的是,铁蛋白抑制剂Fer-1的治疗降低了糖尿病小鼠肾脏中HO-1的水平[18];同样,Kwon等[19]在Erastin诱导的HT-1080纤维肉瘤细胞中观察到HO-1的过表达通过产生过多的Fe2+而引起铁死亡;此外,HO-1基因敲除及药物抑制均证实HO-1的激活通过铁超载和随后产生的过量ROS和脂质过氧化引起铁死亡[11]。因此,HO-1在铁死亡中可能起着复杂的双向调控作用,其具体机制还有待进一步研究。
2 铁在IBD中的作用
铁一般在小肠经主动转运过程被吸收,食物进入肠道后,肠道黏膜细胞内的转铁蛋白在肠腔中与食物中的铁结合,而后与肠黏膜微绒毛上的TFRC结合进入肠黏膜细胞。过量的口服铁在肠道内通过Fenton反应和Haber-Weiss反应产生ROS,从而引发氧化应激,导致一系列的肠道疾病。首先,过量的铁参与Fenton反应引起肠道细胞脂质过氧化;其次会引起胞内线粒体损伤;再次,过量铁诱发内质网应激加重肠道炎症;最后,还能通过减少短链脂肪酸抑制益生菌的生长,破坏肠道菌群平衡[20]。
早期研究认为,IBD可因吸收不良而引起缺铁性贫血,而临床上已将口服铁用于治疗IBD缺铁性贫血,但过量的铁摄入会导致肠道铁超载,引起ROS失调、扰乱肠道菌群而加重IBD。血色素沉着症基因Hfe隐形突变时会出现近端肠道铁吸收增加,Hfe敲除小鼠结肠中丙二醛(malonaldehyde,MDA)升高,更容易出现实验性结肠炎。除此之外,葡聚糖硫酸钠(dextran sulfate sodium,DSS)诱导的实验性结肠炎小鼠的结肠组织中也观察到FTH和铁蛋白轻链(ferritin light chain,FTL)表达增加[21]。这些都强调了铁超载在结肠炎中的病理作用,但存在矛盾的是,之前的研究报道表示,口服铁螯合剂能改善大鼠的实验性结肠炎,但Ettreiki等[22]予幼龄鼠口服铁却预防了其肠道菌群的失调和结肠炎的发生。
3 铁死亡在IBD中对IEC的作用机制
3.1 Gpx4调控铁死亡对IBD中IEC的作用机制Wang等[23]用DSS诱导小鼠发生UC时观察到铁死亡的关键抗氧化酶GPX4 mRNA和蛋白水平受到抑制,而仙茅苷改善了这种抑制和肠道炎症。弗林蛋白酶又称Furin是一种原蛋白转化酶,一篇高质量的实验研究报道了其在CD4+T淋巴细胞中的丢失会引起严重的自发性结肠炎[24],而Dong等[25]在DSS诱导的UC小鼠模型的肠上皮细胞中也发现了Furin水平的明显下调和类似铁死亡的细胞损伤,同时他们观察到Furin不影响ACSL4等铁死亡相关基因的表达,但却对GPX4有明显上调作用,而此机制与激活Nrf2有关。Mayr等[26]在CD模型小鼠中发现,GPX4对PUFAs刺激的肠上皮脂质过氧化具有保护作用,但IEC死亡并不是缺乏一个Gpx4等位基因的小鼠发生肠道炎症的先决条件,他们认为,或许一个Gpx4等位基因足以防止ICE的铁死亡,但不足以防止PUFAs诱导炎症因子的产生,同样在CD患者的结肠组织中也观察到Gpx4水平明显下降。
3.2 Nrf2-HO-1通路调控铁死亡对IBD中IEC的作用机制从上述可知,Nrf2是抑制铁死亡的关键因子,但其过度的激活可能会导致铁死亡。在Chen的研究中,Nrf2和HO-1在DSS诱导的UC小鼠中的表达增加而铁死亡抑制剂Fer-1逆转了Nrf2和HO-1的表达,且明显上调结肠IEC中GPX4和FTH1的表达,抑制环氧合酶2(cyclooxygenase-2,COX-2)和ACSL4的表达,这充分证明铁死亡可能通过Nrf2-HO-1通路对UC上皮细胞发挥作用。黄芪多糖已被证实对UC具有明显的改善作用,而黄芪多糖同样能降低DSS刺激后小鼠结肠组织中Nrf2和HO-1的表达,降低铁死亡相关因子PTGS2、FTH和FTL的表达,并以剂量依赖的形式恢复铁死亡标志物MDA、GSH和铁超载的水平至稳态水平,并且对人类Caco-2细胞铁死亡有抑制作用[21]。去铁胺是一种螯合剂,可用于急性铁中毒的治疗,吴学梅[27]用去铁胺治疗DSS诱导的UC小鼠,发现其不仅能调节炎症因子的释放,还促进结肠组织中Nrf-2通路中相关抗氧化蛋白Keap-1、HO-1的表达来改善炎症。
3.3 内质网应激调控铁死亡对IBD中IEC的作用机制内质网应激已被证实可加重肠道炎症,无论是淋巴细胞还是IEC内质网应激都会引起肠功能紊乱,进而发生或加重炎症性腹泻。选择性抑制内质网应激信号的关键应激传感器—PERK,可明显降低IEC铁死亡,改善实验性结肠炎,而IEC中NF-κB p65的特异性缺失通过内质网应激介导的IEC铁死亡加重了DSS诱导的小鼠UC[5]。
4 讨论
IBD在世界范围内呈上升趋势,尽管其确切的发病机制尚不清楚,但无论是在人类患者还是动物模型的肠道黏膜中,均能观察到广泛而不受控制的IEC死亡,这种死亡可以通过扰乱肠道屏障的紧密连接加重炎症,也会导致肠道免疫系统过度激活、促炎因子、趋化因子过度分泌,从而使肠道黏膜发生继发性损伤,这将是一个恶性循环。
铁死亡是一种新发现的程序性细胞死亡,在肿瘤和神经系统疾病、缺血性/再灌注中已经得到较为广泛的研究,但在肠道研究中,所涉及的内容并不多。例如,在结直肠癌中,Erastin能诱导直肠癌细胞凋亡[28],但其具体机制尚不清楚。在肠道炎症中,ACSL4和PTGS2在结肠炎小鼠的结肠组织中表达均上调[5,29];P53在慢性UC患者中有较高的突变率,P53基因敲除的小鼠肠道内会出现炎症的组织病理学改变;UC患者常因吸收问题而缺乏叶酸,服用叶酸或其代谢前体能缓解结肠炎相关组织损伤[30];然而以上作用是否通过对铁死亡的调控而对肠道炎症有改善或加重和其中的具体机制还有待进一步的研究。但更为直接的是,一些确切强效的铁死亡抑制剂如铁螯合剂、Fer-1和Lip-1可减轻结肠炎相关的肠道损伤,相反,铁、膳食PUFAs等促铁死亡因素则可加重肠道炎症。
更为重要的是,广泛用于治疗中重度IBD的阿达木单抗和英夫利昔单抗却不能改善由口服铁加重的结肠炎小鼠的炎症;5-氨基水杨酸治疗IBD的部分作用是通过上调HO-1发挥的;除此之外,用于治疗UC的抗炎药物磺胺吡啶可通过抑制系统Xc-的功能而诱导铁死亡[1];这些都强烈暗示了调控铁死亡相关途径在治疗IBD方面不可替代的价值。这些在某种程度上强调了铁死亡在IBD中至关重要的作用,但当前的研究远不能阐明其具体机制。除此之外,肠道疾病的铁死亡研究都集中在了IEC上,但在不同环境、不同细胞中同一因素对铁死亡的影响和其机制可能是不同的,而免疫细胞作为IBD发病和进展的关键,是否在肠损伤中发生铁死亡尚不明确。因此,需要更广泛和深刻的研究,以进一步阐明铁死亡在IBD和其它肠道疾病发病机制中的意义。
