RUNX2基因变异致CCD合并严重脊柱侧弯家系分析
2023-12-05夏小雪刘瑞虹黄浩华林斯晓梁红梅
夏小雪 刘瑞虹 黄浩华 林斯晓 梁红梅
1.中山大学附属第五医院口腔科 (广东 珠海 519000)
2.中山大学附属第五医院临床实验医学部 (广东 珠海 519000)
3.珠海固得迎宾北口腔门诊部 (广东 珠海 519000)
4.中山大学附属第五医院中心实验室 (广东 珠海 519000)
锁骨颅骨发育不良综合征(Cleidocranial dysplasia CCD,OMIM: 119600)是一种罕见常染色体显性遗传病[1],发病率为1/1000000。CCD患者典型的临床表现包括发育迟缓,身材矮小,额叶和顶叶隆起,颅骨缝线开放或延迟闭合,锁骨发育不良,肩关节活动度大,双肩下垂等[2]。患病群体中相关的牙齿异常包括乳牙固连、恒牙埋伏阻生[3]、多生牙(70%)、平行的下颌升支[4-6]、反(牙合)、颌骨囊肿等[7]。RUNX家族转录因子2(RUNX2)(基因ID:860, OMIM 600211)杂合性功能缺失突变是导致CCD病例的主要病因。然而,在目前的研究中,基因型与锁骨受累性或胸腰椎侧弯之间是没有明确的相关性报道[2,8-9]。本文详细分析了这两例CCD患者的临床表现,对比临床变异性,旨在提高对本病的认识,减少误诊和漏诊。
1 临床资料
例1:患者李某因牙齿龋坏,2021年8月19日初次于我科就诊,大体检查见:患儿身高106㎝,体重16.4公斤,体脂率14.6%,头围55.5厘米。查体配合,未发现明显的智力障碍。倒梨形头盖骨,颅骨塌陷,前额宽而平坦,头盖骨短而平,额叶隆起,可触及开放的额叶囟门,眼裂向下倾斜,双眼眼距略宽,鼻梁塌陷,面中1/3发育不足。肩关节运动度正常,双肩下垂,脊柱侧弯,手指末端粗短,该患者初诊时面像如图1所示。口腔检查:乳牙列,乳牙广泛龋坏,无明显松动,前牙反(牙合)。口腔黏膜未见明显异常,该患者初诊时口内像如图2所示。初诊时的X光片如图3所示,图3曲面断层片显示下颌骨升支异常,U形乙状切迹,下颌骨正中骨缝未闭。乳牙列,乳牙根未见明显吸收,未萌恒牙在上下颌骨内阻生埋伏,牙根发育不足三分之一,且上下颌前牙区可见埋伏多生牙。图3CBCT显示上颌中切牙舌侧3颗多生牙,下颌侧切牙舌侧各1颗多生牙,共5颗多生牙,左侧下颌第二恒前磨牙形态异常,下颌双侧第一磨牙冠远中。图3胸部CT可见脊柱侧弯,锁骨较正常长度短。手部、脚部CT未见明显异常。结合临床表现,高度怀疑患者为CCD,建议进行基因学检查。患者家长同意进行检测并签署知情同意书。
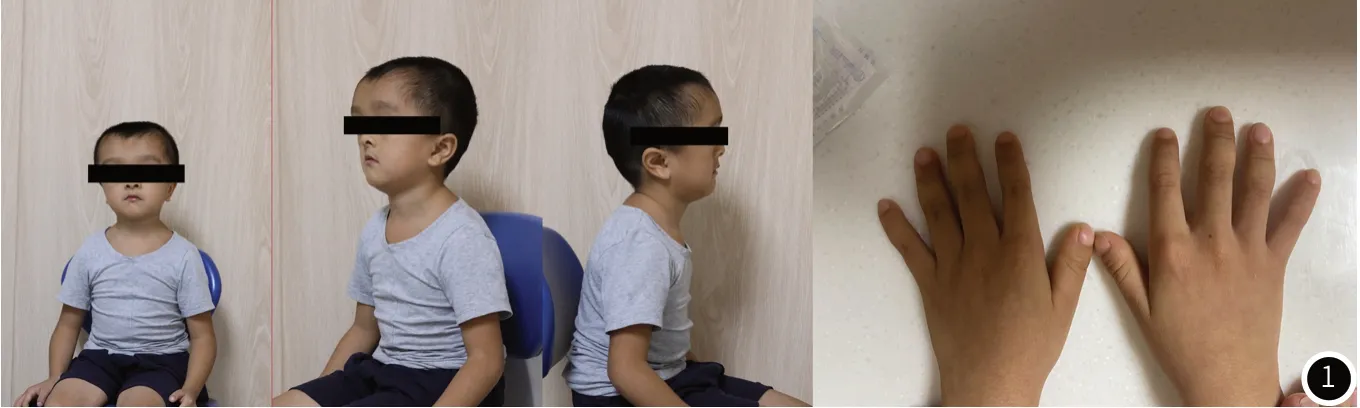
图1 初诊面像。

图2 初诊口内像。

图3 初诊X光片。
2 遗传学分析
使用QIAamp DNA Midi试剂盒(Qiagen,德国)提取该家系中成员的外周血样本中基因组DNA。对该家系的先证者进行全外显子组测序(图4, II-1)。变异致病性评级是根据美国医学遗传学与基因组学学会(ACMG)发布的指南进行相应评估[10]。全外显子组测序结果显示患儿在RUNX2 (NM_001024630.3)基因中存在一个罕见的杂合无义突变c.1096G>T (p.Glu366*)。Sanger测序来验证获得的候选变异以及对其他患病家系成员(I-2)的变异的确认来进行家系基因型-表型共分离分析 (图4)。结果显示,患病的母亲在RUNX2中也发现了相同的突变。开放阅读框预测发现该突变在肽链366位提前引入终止密码子,使3’端156个氨基酸被截断(ORF Finder预测),丢失了整个Runx抑制结构域(Runx inhibition domain) (图4), https://www.ebi.ac.uk/interpro/protein/UniProt/Q13950/entry/pfam/#table)。MutationTaster预测此突变还会引起无义突变介导的mRNA降解 (nonsense-mediated mRNA decay, NMD) (https://www.mutationtaster.org/),导致携带该突变的等位基因没有实际上的表达。根据ACMG指南,该突变可归类为“致病性变异”[10]。因此,遗传学诊断是:颅骨锁骨发育不良综合征。口腔诊断是:安氏III类错颌畸形,龋齿,多生牙,恒牙迟萌。结合口内情况,建议患者行正畸埋伏牙牵引治疗,因患者于外地上学,就诊时间不便,先行脊柱侧弯保守治疗,暂不进行口腔科相关治疗。患者再次就诊时间为2023年1月20日,此时患儿口内情况如图5所示,乳牙列,口腔卫生差,牙结石I度。左侧上颌乳中切牙、侧切牙缺失,乳牙多发龋齿,下颌乳中切牙、右侧上颌乳中切牙、第一乳磨牙II度松动。X光片如图6所示,图6曲面断层可见预估上下颌中切牙牙根发育至三分之二,下颌双侧第一磨牙远中阻生较之前严重。同时图5CBCT显示右侧下颌尖牙区一迟发性多生牙。结合患儿口腔内情况及图6头颅侧位片可见,患儿反(牙合)情况加重。新增诊断:迟发性多生牙。给出新的治疗方案,建议行埋伏牙正畸牵引的同时进行反(牙合)功能矫治。于2023年3月22日行上下颌乳前牙拔除术+上下颌切牙开窗粘结牵引装置+多生牙拔除术+下颌第一磨牙开窗粘结牵引装置+上颌第一磨牙开窗术。术中发现上颌中切牙及下颌第一磨牙牙面较其他恒牙色黄,表面粗糙,高度怀疑存在牙釉质发育不全。现下颌磨牙已萌出,余下牙齿正畸牵引(牙合)向移动,反(牙合)改善。
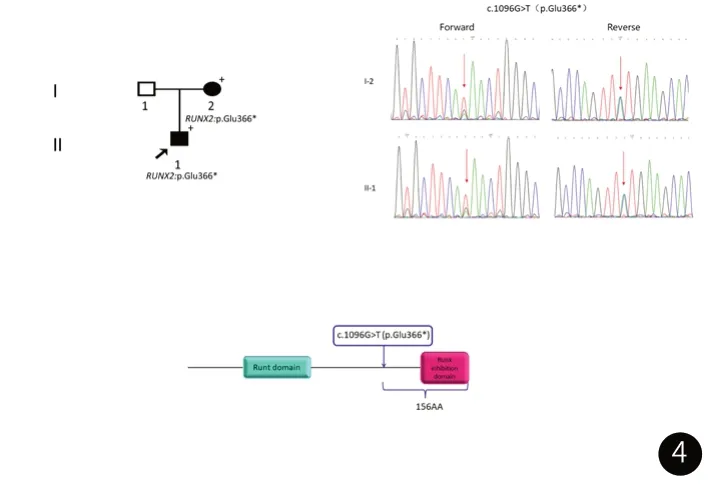
图4 CCD家系的遗传分析家系图。+表示可用于基因分析的个体。Sanger测序图。结构域图。野生型RUNX2蛋白含有一个runt结构域(蓝色)和一个runt抑制结构域(粉红色)。箭头表示突变的位置(c.1096G>T, p.Glu366*)。

图6 复诊X光片。
例2:为先证者母亲,患者李某,女性,36岁。与例1存在相似的临床表现,但无脊柱侧弯。该患者初诊时面像如图7所示。口腔检查:上颌见活动义齿,固位差,下颌冠桥修复体(#36-#46),垂直距离低。口腔黏膜未见明显异常,该患者初诊时口内像如图8所示。初诊时X光片如图9所示,图9曲面断层片显示下颌骨升支异常,U形乙状切迹。恒牙列,上颌为无牙颌,下颌#36、#35、#41、#45、#46骨内埋伏阻生,冠根发育基本正常,上下颌前牙区可见大量多生牙。多生牙部位及个数与例1相同,但无异形牙。图9胸部CT可见锁骨较正常长度短,手部、脚部CT未见明显异常。因患者儿子已确诊为CCD,结合患者临床表现,高度怀疑患者同为CCD,建议进行基因学检查。Sanger测序证实该患者与其子一样携带RUNX2基因的杂合性致病性突变。遗传学诊断是:颅骨锁骨发育不良综合征。口腔诊断是:上牙列缺失,下牙列缺损,下牙列不良修复体,多生牙,阻生齿。当患者再次就诊时下颌冠桥不良修复体II度松动,无法咀嚼硬物,上颌活动义齿固位差。此时患者X光片如图10所示,图10曲面断层可见#36、#45、#46根尖周阴影。结合患者经济状况,给出治疗方案,拔除#36、#35、#41、#45、#46,拟采用上下颌活动义齿恢复颌间距离与咬合关系。现已重新进行义齿修复。

图7 初诊面像。
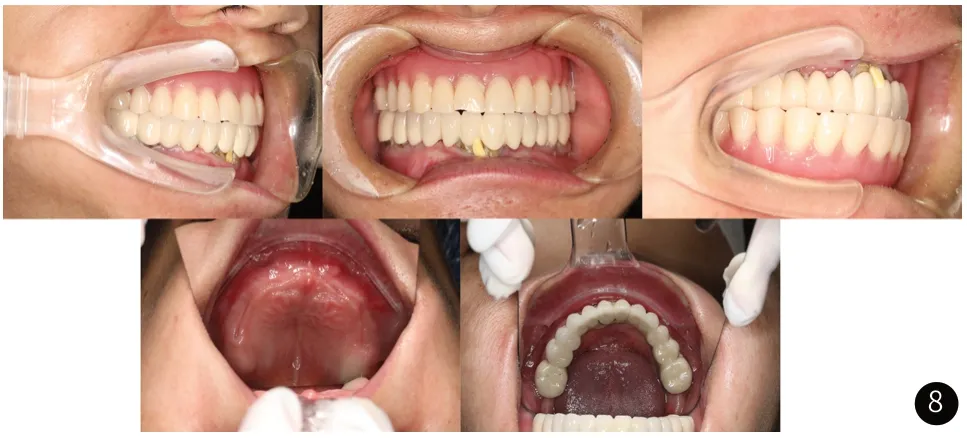
图8 初诊口内像。
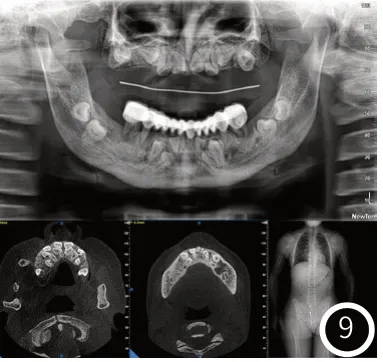
图9 初诊X光片。

图10 复诊X光片。
3 讨 论
CCD患者的口腔常表现为乳牙滞留、恒牙列迟萌、数目不等的多生牙、咬合异常等错颌畸形[11],口腔科医生应了解CCD的主要特征,避免误诊,当患者临床症状或者放射学上表现不明显时,可考虑遗传学分析,方便发现家族性的其他病例,便于早期治疗。当发现患者存在CCD特征性的临床表现及影像学表现时,基本上可以进行临床确诊,但明确诊断还需进行基因检测。CCD患者口腔治疗过程较为复杂,治疗周期长。需正畸科、牙体牙髓病科、颌面外科、修复科等多学科联合治疗。其治疗原则是:尽可能地做到早诊断、早干预、早治疗,这样不仅可以做到尽早对患者因病施治,减轻牙颌畸形程度,降低后期治疗难度,也能够最大程度地满足患者对颜面美观的要求。例1中患儿发育迟缓,身材矮小,考虑到恒牙牙根及颌骨发育情况,主要参考Jerusalem法[12],一期治疗为了暴露未萌恒切牙,需将 CCD 患者的乳前牙和多生牙全部拔除,并在开窗暴露的恒切牙上粘接正畸牵引装置,术区利用周围软组织皮瓣缝合关闭。同时,因CCD患者的上颌骨体较小,上颌牙弓狭窄及牙列反(牙合)的问题必须优先纠正[13],需配合上颌扩弓+前方牵引。二期治疗预计在两年后,需再次配合外科手术,拔除剩余乳牙,去除阻生恒牙萌出通道的骨阻力,以利于阻生牙正常萌出。开窗暴露未萌尖牙和前磨牙,需进一步正畸牵引[14-15]。此阶段根据颅面骨的发育情况,继续配合功能矫治。考虑到例1中患儿颌骨发育情况,患儿成年后极大可能需要配合正颌手术解决骨骼畸形,从根本上改善面型,实现颅颌面整体协调和美观。针对CCD患者的正颌手术干预,应以恢复患者颅颌面的功能和美观为最终目的,根据其年龄、个人需求和经济条件等因素综合考量,制定矫治方案[16-18]。对于患有CCD的成年人,应始终考虑修复前正颌手术,以纠正上颌发育不足,再行种植或者修复治疗。例2考虑到患者的诉求及经济状况,放弃正颌手术,保留全部埋伏牙及多生牙,维持现有牙槽骨高度及宽度,通过上下颌活动义齿进行咬合重建,恢复咀嚼功能,改善面容。CCD的骨骼、颅颌面部异常复杂程度增加了正畸治疗的难度和不确定性,治疗方法仍在探索中。
RUNX2基因功能缺失杂合性突变主要可引起具有典型牙齿表现的CCD。然而,RUNX2基因的基因型与脊柱侧弯的表型之间没有明确的相关性报道[6,8-9]。本研究中c.1096G>T (p.Glu366*)突变曾在一个中国家系的两名患者中被报道,他们也表现出牙齿典型症状,但并没有出现任何脊柱侧弯的临床表型[19]。在本研究中,我们首次报道了该变异与胸腰椎侧弯表型相关。并且,先证者的外显子组测序结果也排除了他携带其他已知导致脊柱侧弯的基因致病突变的可能性。因本文病例1中乳牙大量龋坏,无法判断是否存在牙齿釉质、牙本质矿化不足。外科开窗手术中发现上颌中切牙及下颌第一磨牙牙齿牙面色黄,质地不均匀,待牙齿牵引萌出后,才可判断。本文中母子基因突变相同,但临床表现存在不同,有可能该病还有其他因素影响其表现度,需进一步研究探讨。儿子与母亲存在同样数目及部位的多生牙外,发现一颗迟发性多生牙,且#35为异形牙。并伴有严重的脊柱侧弯。儿子较母亲异常情况更多。母亲临床表现可视为儿子不进行治疗干预后临床表现自然转归,而且更加严重,提示我们CCD早期治疗干预的重要性。
