血源性人凝血因子Ⅷ治疗血友病A 患者的有效性及安全性评价
2023-11-30梁舒敏张伟蒋桂香孙忠良郑炎闫晨
梁舒敏 张伟△ 蒋桂香 孙忠良 郑炎 闫晨
(1.广东双林生物制药有限公司,广东 湛江 524000; 2.上海凌先医药科技有限公司)
健康人发生出血时,凝血因子Ⅷ与活化的凝血因子Ⅸ、钙离子、磷脂形成复合物,辅助激活凝血因子Ⅹ,实现内源性凝血。 而血友病是一组性联隐性遗传的出血性疾病,患者由于缺乏凝血因子Ⅷ或凝血因子Ⅸ,导致血液不能正常凝固,引起不受控出血,临床上血友病主要分为血友病A 和血友病B 两类,血友病A 以凝血因子Ⅷ缺乏为主;血友病B 以缺乏凝血因子Ⅸ为主,其中血友病A 约占血友病患者总数的80%以上,男性发病率为1/5 000[1]。 1986—1988 年全国24 省市37 个地区对血友病进行调查,初步查明中国血友病的患病率为2.73 人/10 万人口[2],截止2021 年5 月,中国约有10 万血友病A 患者,其中接收治疗登记在册的患者病例36 049例[3],未接受治疗的人群基数仍较大,而输注人凝血因子Ⅷ的替代疗法是治疗血友病A 的主要手段。 因FⅧ在机体内的半衰期仅为8~12h,止血效果虽然显著但不能持久,再次出血时需反复输注[4]。 故血友病A 患者需要终生使用FⅧ制剂进行替代治疗,长期来看,无论是按需治疗或是预防治疗,FⅧ制剂的需求量都将持续增加。 随着蛋白分析、纯化及病毒灭活技术的不断提高,血浆来源的高纯度FⅧ制剂,被广泛用于治疗和预防血友病A 患者的自身出血和术后出血等领域。 本研究由国内5 家中心共同完成,旨在评价一种血浆源性FⅧ制剂治疗血友病A 的有效性和安全性。
1 材料与方法
1.1 药品 人凝血因子Ⅷ,规格:200 IU/瓶,外观成乳白色疏松体,复溶后为无色澄明液体,可带轻微乳光。 本品临床试验批件号为2015L03019,本临床试验分别在济南市中心医院、山东大学第二医院、河北医科大学第三医院、郑州人民医院、潍坊医学附属医院等5 家研究单位开展(临床研究注册号:CTR20160129)。
1.2 用量 本研究中FⅧ用量由研究者根据受试者体重,出血的严重程度等因素,定制给药方案;FⅧ用量计算公式为:所需FⅧ单位(IU)/次=0.5×受试者体重(kg)×计划提升的FⅧ活性水平(%)。 其中受试者“计划提升的FⅧ活性水平(%)”需根据出血程度来确定,具体见表1。

表1 出血程度及预期提升FⅧ活性水平(%)
1.3 病例选择
1.3.1 纳入标准 1)年龄≥6 岁且≤65 岁;2)临床确诊为血友病A,伴有自发性出血或外伤出血的临床表现;3)患者曾接受过凝血因子Ⅷ的替代治疗;4)所有生育年龄的受试者都必须在进入筛选期后直至研究完成后3 个月内采取有效的避孕措施;5)成年患者自愿签署知情同意书,未成年受试者经其法定监护人同意并自愿签署知情同意书,组长单位济南市中心医院医学伦理委员会临床试验伦理批件号:济中心伦理临审2016004 号。
1.3.2 排除标准 1)确诊为除血友病A 之外的其它出血性疾病;2)对凝血因子Ⅷ制剂任何成分和其他蛋白类血液制品过敏者;3)严重的心脑血管疾病,包括心肌梗死、心功能不全3 级以上者;或有血栓病史等其他严重的疾病、以及未受控制的全身疾病,研究者认为不适宜者;4)受试者在进入研究后的6 个月内可能接受择期大中型手术;5)已经出现或曾经出现凝血因子Ⅷ抑制物阳性(定义为大于实验室正常值上限,一般Bethesda 法≥0.6 BU/mL);6)肝功能(ALT、AST、TBIL)≥2 倍正常值上限或肾功能(BUN、Cr)≥1.5 倍正常值上限;7)病毒检测HBsAg 阳性、抗-HCV 阳性、抗-HIV 阳性、梅毒螺旋体抗体阳性;8)受试者正在接受基础性的凝血因子Ⅷ预防性治疗;9)受试者入组前3 d 内使用过任何凝血因子Ⅷ制剂;10)入组前3 个月内接受过成分输血或研究期间可能需要成分输血的受试者;11)研究期间需要使用抗凝或抗血小板治疗的受试者;12)入组前1 个月内参加过其他药物临床试验的受试者;13)孕妇或处于哺乳期的妇女;14)其他任何研究者认为不适合参加本临床试验者,包括无法或不愿意遵守试验方案的要求者。
1.4 评价标准
1.4.1 主要疗效指标——FⅧ活性输注效率值 根据出血状况不同,通过计算输注结束后10 min FⅧ活性输注效率值,以比较药物的输注效果。 FⅧ活性输注效率值的计算公式如下:
1.4.2 次要疗效指标——临床总有效率 输注后24 h 对所有受试者进行出血症状和体征改善评判,计算临床总有效率。 具体评判标准见表2。 总有效率的计算公式如下:

表2 出血症状和体征改善评判标准
1.4.3 安全性评价指标 1)记录体温、心率、呼吸、血压。 2)记录心电图;治疗前后检查血常规、肝肾功能;筛选及治疗后d90 和d180 检查HBsAg、抗-HCV、抗-HIV、梅毒螺旋体抗体、凝血因子Ⅷ抑制物。 3)记录不良反应及不良事件:不良反应是指临床试验中的受试者出现的与试验药物有关的不良事件或者严重不良事件。 不良事件(adverse event, AE):是指受试者在临床试验过程出现的不良医学事件,不良事件不一定与试验药物间有因果关系。 严重不良事件(serious adverse event, SAE):是指临床试验过程中发生需住院治疗、延长住院时间、致伤致残、影响工作能力、危及生命或死亡、导致先天畸形、死亡等事件。 重要不良事件(significant adverse event):指的是除严重不良事件外,发生的任何导致采用针对性医疗措施(如停药、降低剂量和对症治疗)的不良事件和血液学或其他实验室检查明显异常。 本研究只统计采取治疗措施的不良事件。
1.5 统计分析方法 本研究数据采用SAS 9.3 软件进行分析。 对于主要疗效指标,计算其均值、标准差、Q1 ~Q3、最小值、最大值以及中位数。 对于次要疗效指标,均以频数表、百分率描述。 主要疗效指标及次要疗效指标分析同时采用全分析集(FAS)和符合方案分析集(PPS),其中未纳入PPS 的4 例受试者均已完成主要疗效指标及次要疗效指标数据收集,对疗效指标分析有统计学意义,故本研究以FAS 为主要分析集,安全性分析采用安全性数据集(SS)。
1.6 伦理学 在研究开始前,研究者已将临床试验方案、患者知情同意书以及本研究相关的资料递交各研究中心的EC审批,并获得了实施该临床研究的批准文件,批件号为济中心伦理临审2016004 号。 EC 的批准文件均以书面形式递交研究者,在研究过程中,严格按照EC 要求落实各项工作,在执行修订的临床试验研究方案前,均已通过EC 审批并获得相应批准。
2 结果
2.1 受试者一般资料 本临床试验选择了6 家研究中心,启动5 家,共筛选69 名受试者,入组54 名受试者,52 名受试者完成整个研究。 本研究54 名受试者进入SS 集与FAS 集,其中入组的4 名受试者分别因受试者本人要求退出试验、试验期间输注红细胞、拒绝随访、合并用药等情况,而未进入PSS集,合计50 名(92.59%)受试者进入PPS 集(表3)。
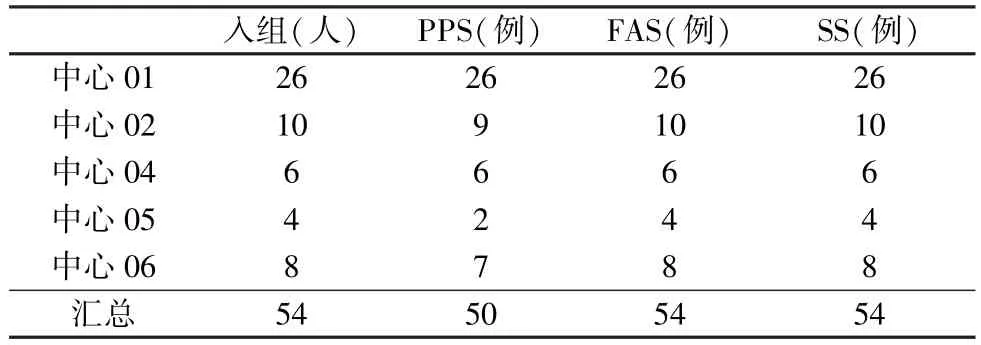
表3 受试者与病例数据集分布
FAS 集人群中,受试者平均年龄为26.9 岁,中位年龄为26.0 岁,受试者最大年龄与最小年龄分别为53 岁与11 岁;受试者平均体重为(66.38±15.40)kg;平均身高为(172.1±6.35)cm;受试者用药前的FⅧ活性基线中位值为1.60%,最低值为0.7%,最高值为4.3%;54 名受试者中,血友病临床分型为中型(FⅧ:C 1%~5%)的患者50 人,重型(FⅧ:C<1%)患者4 人(表4)。 受试者的基线体温、心率、呼吸、血压等生命体征平稳;54 名受试者均为男性;受试者总体依从性良好。FAS 的54 名受试者均为血友病A 的患者,其中29 名(53.7%)受试者有血友病A 家族病史。 5 名(9.3%)受试者有除血友病外其他病史。

表4 受试者一般情况汇总
2.2 受试者首次出血事件试验药物暴露剂量 在FAS 中,受试者首次输注用量均值为(955.6±247.76)IU,中位值为1 000 IU;受试者首次输注每公斤体重用药量均值为(14.376 0±1.665 9)IU,中位值为14.626 IU(表5)。
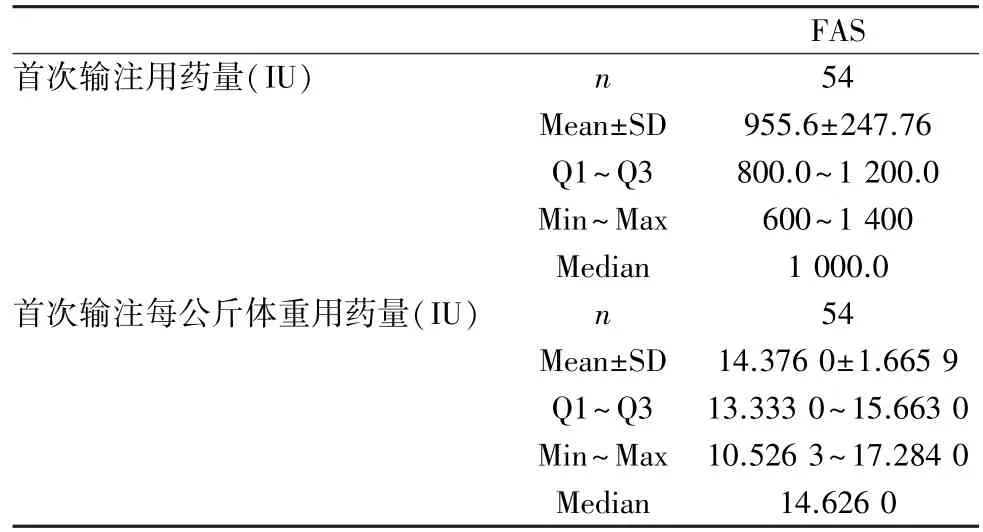
表5 首次出血事件试验药物暴露情况
2.3 疗效结果
2.3.1 主要疗效指标结果 在FAS 中,患者FⅧ活性基线值均值为1.69±0.735,中位值为1.60%,首次输注后10 min 的患者FⅧ活性实测值较基线值升高幅度均值为(49.18±13.071)%,中位值为47.35%,PPS 的结果与FAS 基本保持一致(表6)。 FⅧ活性输注效率值的目标值为100%,FAS 中,受试者首次输注后10 min 的FⅧ活性输注效率值均值为171.9%,中位数为169.5%,比较可知,首次输注后10 min 的FⅧ活性输注效率值的均值和中位数均高于设定的目标值100%,且PPS 的结果与FAS 基本保持一致(表7),所以FⅧ在首次输注后10 min 能获得很好的FⅧ活性输注效率值。各分中心结果相似,PPS 的结果与FAS 基本保持一致。 由此显示,FⅧ在首次输注后能获得良好的FⅧ活性输注效率。
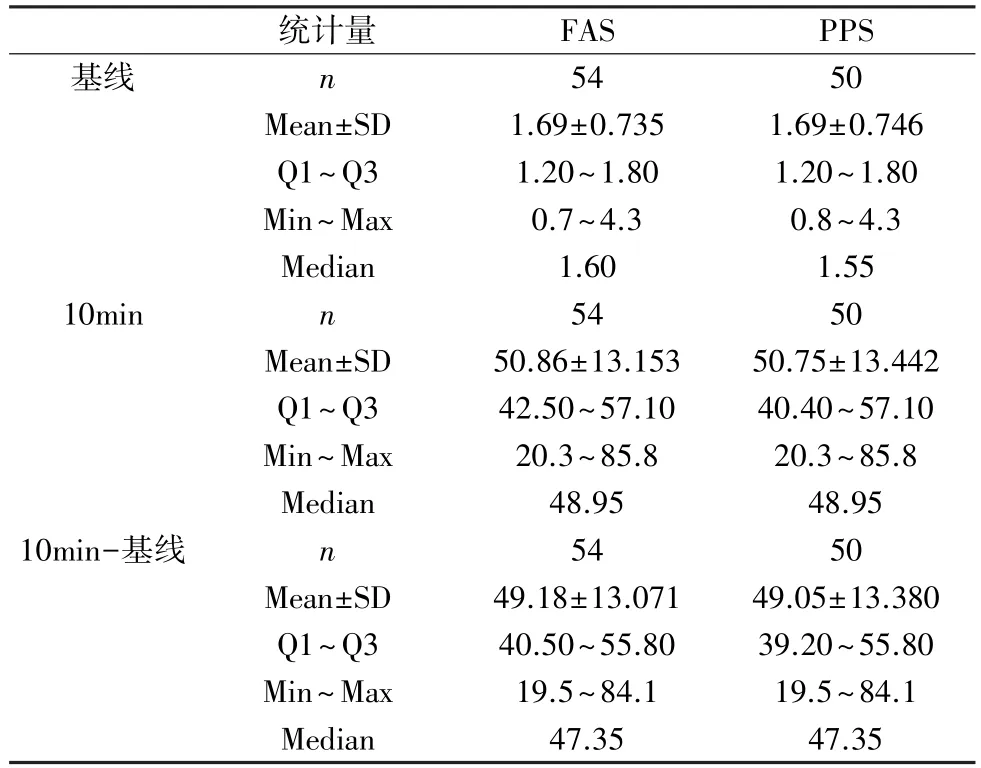
表6 首次输注凝血因子Ⅷ活性实测值和变化值(%)分析情况(FAS/PPS)
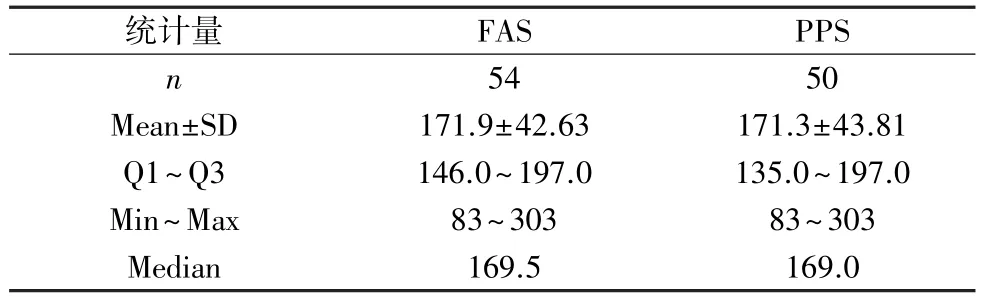
表7 首次输注后FⅧ活性输注效率值
2.3.2 次要疗效指标结果 在FAS 中,在首次输注后24 h内对受试者出血症状与体征改善进行评分,其中“显效”19例(35.2%),“良效”35 例(64.8%),根据统计学四分位方法分层,每公斤体重用药量为Q1 以下的患者合计13 名,其中“显效”2 例(15.4%),“良效”11 例(84.6%),每公斤体重用药量为Q1 ~Q3 的患者合计28 名,其中“显效” 14 例(50.0%),“良效”14 例(50.0%),每公斤体重用药量为Q3 以上的患者合计13 名,其中“显效”3 例(23.1%),“良效”11 例(76.9%),所有患者均达到良好的替代治疗效果。 输注后24h 内出血症状与体征的临床总有效率达到100%。 PPS 的结果与FAS 基本保持一致(表8),表明人凝血因子Ⅷ在首次输注后24 h 内能控制患者的出血症状,临床症状改善明显。在FAS 中,根据统计学四分位方法分层,单位体重用药剂量为Q1 以下的患者合计13 名,其输注效率值均值为171.1±37.48,单位体重用药剂量为Q1~Q3 的患者合计28 名,其输注效率值均值为174.8±46.0,单位体重用药剂量为Q3 的患者合计13 名,其输注效率值均值为166.4±42.45,从结果判断,不同单位体重用药剂量其输注效率值评定情况相近,PPS的结果与FAS 基本保持一致(表9)。 由此说明,FⅧ在首次输注后,FⅧ活性较基线有明显的提升;不同单位体重用药量下,其输注效率值相近,临床总有效率均为100%。
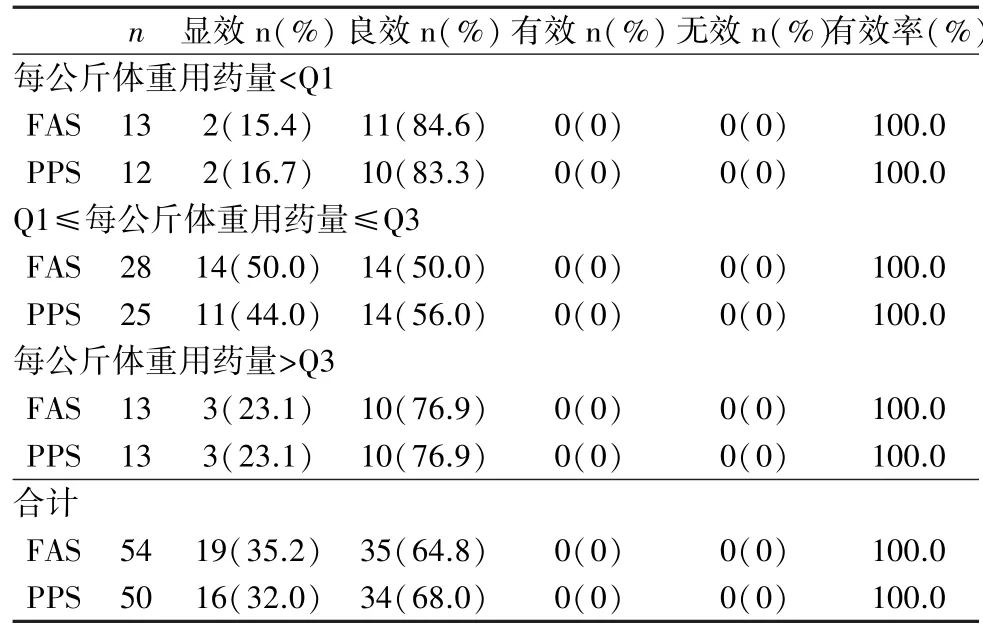
表8 首次输注不同单位体重用药量分层的出血症状和体征改善评定情况(FAS/PPS)

表9 首次输注不同单位体重用药量分层的恢复率情况(FAS/PPS)
2.4 安全性评价
2.4.1 安全性分析集 安全性评价使用安全性数据集(SS)。至少输注过一次研究药品,且有安全性指标记录的实际数据的受试者进入SS。 本次研究共入组受试者54 名,全部进入SS。
2.4.2 不良事件 在研究中,用药后共5 名(9.3%)受试者发生6 例次与药物相关的不良反应,分别为发热2 例(3.7%)、高热1 例(1.9%)、寒战1 例(1.9%)、血胆红素升高1 例(1.9%)和恶心1 例(1.9%),均为轻度不良反应,未发生中度和重度不良反应,无导致脱落的不良反应发生。
在研究中,治疗前共28 名(51.9%)受试者病例发生不良事件,用药后共38 名(70.4%)受试者发生96 例次不良事件,无导致脱落的不良事件发生。 用药后96 例次不良事件按SOC 分类,其中感染及侵染类疾病共19 例(35.2%),各类实验室检查共16 例(29.6%),各种肌肉骨骼及结缔组织疾病共6 例(11.1%),全身性疾病及给药部位各种反应共6 例(11.1%),胃肠系统疾病共6 例(11.1%),其余发生率均较低。 用药后共38 名受试者中,共32 名(59.3%)受试者发生48 例次重要不良事件,无导致死亡的不良事件;4 名(7.4%)受试者发生4 例次严重不良事件,分别为外伤性出血1 例、外伤性血肿1 例、缺铁性贫血1 例、失血性贫血1 例,均为出血症状的表现,为血友病的主要症状表现,以上不良事件经研究者判断,与研究药物无关。
2.4.3 实验室检查 本研究中,受试者接受体温、心率、呼吸、血压、身高和体重等各项生命体征指标检查。 试验期间,生命体征指标无明显变化。 本研究实验室检查中,输注前正常输注后异常有临床意义的病例如下:红细胞计数异常降低4 例、淋巴细胞百分比异常增高1 例、血红蛋白异常降低3 例、尿红细胞异常增高1 例、尿白细胞异常增高2 例、尿潜血异常1 例、谷草转氨酶异常降低1 例、总胆红素异常增高1 例、白蛋白异常降低4 例。 其中红细胞计数异常、血红蛋白异常、尿红细胞异常、尿白细胞异常、尿潜血异常均为血友病患者出血症状的实验室表现。 输注试验药物后,受试者淋巴细胞百分比为52.1%(正常值范围:20%~50%),略超出正常值范围上限,为轻微升高,谷草转氨酶检查结果为14.9 IU/L(正常值范围:15 ~40 IU/L),略低于正常值范围下限,为一过性轻微降低,研究者出于谨慎考虑,均评估为有临床意义。心电图中,输注前正常输注后异常的病例有8 例,经研究者分析判断,以上实验室数据异常与研究用药无关。
2.4.4 病毒学检查 54 例受试者输注凝血因子Ⅷ治疗后,有53 名受试者完成90 d 随访,其中52 名受试者完成180 d随访,本研究中,在筛选期d90 和d180 进行病毒学指标的监测,病毒学检测(HBsAg、抗-HCV、抗-HIV、梅毒螺旋体抗体)结果均为阴性,无病毒学检测结果为阳性的受试者,凝血因子Ⅷ抑制物均未观察到阳性的病例。
3 讨论
本研究共入组患者54 名,受试者首次输注后10 min 的FⅧ活性输注效率均值为171.9%,中位数为169.5%,均高于设定的目标值100%。 首次输注后24 h 内对受试者出血症状与体征改善进行评分,其中“显效”19 例(35.2%),“良效”35 例(64.8%),临床总有效率达到100%。 5 名(9.3%)受试者发生6 例次与药物相关的不良反应。 治疗后d90、d180 检测HBsAg、抗-HCV、抗-HIV、梅毒螺旋体抗体、凝血因子Ⅷ抑制物,未发现阴性转阳性病例。 说明本研究中的血源性FⅧ制剂,输注后能在短期内显著提高血友病A 患者的FⅧ活性水平。 而较高的输注效率可以一次性阻断出血的发生,治疗效果显著,能及时有效的控制和缓解出血症状和体征,总体安全性良好。
血源性FⅧ(pFⅧ)和重组FⅧ(rFⅧ)制剂已广泛用于血友病A 的替代治疗。 两者在开展凝血因子替代疗法治疗一段时间后,患者血液中都可能产生凝血因子Ⅷ抑制物。 有报道重型血友病A 抑制物发生率为20%~30%,中型或轻型血友病A 患者中5%~10%也会产生抑制物[5]。 因rFⅧ缺乏vWF 因子,rFⅧ导致患者体内抑制物的产生率为pFⅧ的两倍,相比之下,pFⅧ同源性较高,临床使用也更为安全[6-7]。此外,儿童患者首次治疗时多应用pFⅧ,以降低抑制物的发生。 虽然rFⅧ在国外已应用得比较成熟,但也无法全面替代pFⅧ。
近年来“基因治疗”研究受到社会关注。 目前研究中大多以腺相关病毒(adenoassociated virus,AAV)为载体,但AAV载体易因肝细胞分裂而被稀释,导致治疗效果减弱[8]。 目前临床试验仅用于成年患者[9],在同类产品临床试验中发现,基因疗法最主要的不良事件是肝酶升高[10]。 据报道,全球有约12%(26/217)的国家开展了相关临床试验[11]。 通过基因治疗,部分患者在单次治疗后体内FⅨ或FⅧ活性水平达到正常值[12]。 从现阶段研究进度来看,基因疗法的研究进度较为缓慢,单次治疗后2~3 年仍有表达下调,致使体内FⅨ或FⅧ活性水平逐步降低至较低水平,此外基因疗法价格昂贵,是否能完成替代传统的替代疗法,尚待考究。
此外,非因子类产品(双特异性单克隆抗体)也是研究热点,其中罗氏公司生产的Hemlibra 为重组人源化的IgG4 双特异性单克隆抗体,通过桥接FⅨa 和FⅩ,促进凝血酶的生成恢复A 型血友病患者的凝血过程,达到止血效果。 该药于2018 年11 月获得国家药品监督管理局(NMPA)批准,用于存在凝血因子Ⅷ抑制物的A 型血友病成人和儿童患者的常规预防性治疗,是目前首个可每周一次进行皮下注射的预防性治疗药物。 Hemlibra 的主要不良反应,包括注射部位反应、头痛和关节痛等,而血栓事件风险,更值得关注。
血友病的管理在不断完善,对于血友病的治疗方法研究也进入前所未有的黄金时代,越来越多的新产品逐渐涌现,新方法的出现也引领者pFⅧ及rFⅧ生产厂家不断优化自身产品,如何降低FⅧ的免疫源性,提高FⅧ的半衰期,降低患者治疗频率,提高治疗依从性,也是广大科研工作者需要考虑的问题。
利益冲突:所有作者均声明不存在利益冲突。