炭载单原子催化剂在电还原二氧化碳领域的研究进展
2023-11-21张艺严陈熙元董灵玉郝广平李文翠
张艺严,陈熙元,董灵玉,贺 雷,郝广平,李文翠
(大连理工大学 化工学院,辽宁大连 116024)
自工业革命后,人们对化石燃料的使用需求日益增长,因而大量的二氧化碳(CO2)被排放,导致了温室效应、全球气候变化等一系列环境问题[1,2]。除此之外,化石燃料的有限性和不可再生性也不利于可持续发展。利用可再生能源对CO2进行转化和利用,将其还原成低碳燃料和有经济效应的化工产品,不仅能减少环境中CO2的总量,维持全球碳平衡,还能够有效缓解能源短缺的问题,变废为宝[3,4]。电化学还原CO2技术是目前众多CO2转化技术中备受关注的一种,它具有温和的操作条件、理想的转化效率、可控的反应过程,且能够将可再生电能转化为化学能,实现清洁能源的转化利用和高效存储[5-7]。
电催化CO2还原是利用电能在水溶剂或非水溶剂中使CO2通过通电装置,在阴极的催化剂表面被电还原,发生复杂的电化学反应后生成一氧化碳或其他有机产物的技术。CO2分子对称性高,化学性质稳定,其C=O 键能高达750 kJ/mol。因此,还原反应需要的活化能较高,需要施加较高的负电势(CO2还原反应平衡电位可达-0.47 V vs.SHE[8])来驱动反应。同时,在水溶液中反应时,存在阴极的竞争反应— 析氢反应(Hydrogen Evolution Reaction,HER),随着反应电压的升高,副反应也随之加剧,抑制了CO2的还原,影响了最终的产物分布。因此,合理设计催化剂结构,降低CO2还原活化能,抑制析氢反应,是实现高效ECR 的关键。
“单原子催化”的概念自2011 年由张涛院士团队提出后[9],在催化领域的研究日益广泛。由于催化剂暴露的活性位点数直接影响着催化剂的本征活性,因此,将催化剂减小到纳米颗粒级别甚至原子级别,催化剂的性能会得到明显提升[10]。单原子催化剂(Single-atom Catalysts,SACs)是由原子级别的金属分散于载体基底上而形成的具有单一催化活性中心的新型催化剂,具有高比活性、100%原子利用率和独特的电子结构[11]。近年来,在电催化还原CO2领域,单原子催化剂的研究已成为热点和前沿问题。单原子催化剂的性能与其局域结构和电子性质密切相关,因此,载体起到了重要作用。载体不仅为单原子活性组分提供了支撑和分散的基底,其几何结构和表面性质在很大程度上影响着单原子催化剂的催化性能。
由于单原子的配位环境处于高度不饱和状态,且反应过程中被吸附在单原子上的反应物会减弱单原子与载体之间的相互作用,导致单原子更容易在载体表面发生迁移,形成团簇[12]。因此,不少学者研究了各种不同的合成策略来提高单原子稳定性,总体上分为两大思路:一是通过物理方法(如空间约束、缺陷捕获等)实现金属前驱体的原子级分散和隔离,限制其在载体上的迁移和团聚;二是通过增强单原子与载体之间的相互作用或单原子与周围配位原子直接的电荷转移效应,从而提升稳定性[13]。在炭材料或者金属氧化物等载体中,金属单原子可以与其他原子共享或转移电子,形成化学键,将它们锚定在载体上,化学键的能量决定了单原子与载体之间相互作用力的强弱。此外,单原子与载体之间的相互作用[14]也可能涉及到静电相互作用、范德华力等其他较弱的作用形式。这些较弱的相互作用也会影响单原子催化剂的性质。
目前,单原子催化剂的制备方法有物理方法和化学方法[15],以后者为主。物理方法包括质量分离-软着陆[16]、球磨[17]、微波辅助[18]等;化学方法有“自下而上”和“自上而下”两种不同策略[19-21]。表1列举了一些SACs 常见的制备方法及优缺点。

表1 单原子催化剂的制备方法及优缺点Table 1 Common preparation methods advantages and disadvantages of SACs
对于单原子催化剂来说,证明孤立单原子的存在和明确它们的空间分布是极其重要的,因此,单原子催化剂的表征技术是影响单原子催化剂发展的因素之一[22]。目前,采用较多的表征方法包括探针分子红外光谱[23]、球差校正的高角度环形暗场扫描透射电子显微成像(Spherical Aberration Corrected -High-Angle Annular Dark Field -Scanning Transmission Electron Microscopy,AC-HAADFSTEM)[24,25]、扫描透射电子显微成像(Scanning Transmission Electron Microscopy,STEM)[26]、X 射线吸收光谱(X-ray Absorption Spectroscopy,XAS)等,并结合X 射线光电子能谱(X-ray Photoelectron Spectroscopy,XPS)[27,28]、原子力显微成像(Atomic Force Microscope,AFM)、Kelvin 探针力显微成像(Kelvin Probe Force Microscope,KPFM),扫描隧道显微成像(Scanning Tunneling Microscope,STM)等辅助技术获取活性原子的空间分布、电子结构、配位环境、化学价态等重要信息[29,30]。
单原子电催化还原CO2的反应机制与催化剂的几何构型,电子特性与中间体的吸附方式之间存在内在关系[31]。例如,当金属单原子转化为金属离子(M+)作为反应的活性位点,能够高效活化CO2分子[32];当单个铁原子被载体中的N 和O 共同锚定形成配位结构后,能够促进*COOH 的生成(质子化),优化*CO 的结合强度(解吸),从而促进还原产物CO 的生成[33]。
炭材料包括零维碳点/碳球、一维碳纳米管/碳纳米棒、二维石墨烯、三维氮掺杂多孔炭、石墨炔等,此外,以金属有机框架为前驱体制备的炭材料及过渡金属碳化物(Mxene)等都满足可以作为单原子催化剂载体的要求。本工作根据金属单原子催化剂载体的不同进行分类,综述了近年来不同维度炭材料负载单原子催化剂的制备方法、结构特点及在ECR 领域应用的研究进展,期望能为相关领域的研究提供借鉴。
1 电催化二氧化碳还原炭载体的类型
单原子催化剂的载体可大致分为两类:一类是以金属氧化物作为载体;另一类是以非金属作为载体。金属氧化物作载体是利用分散在载体上的原子与氧原子的强相互作用来锚定单原子[34];非金属作载体则是通过金属原子与载体上的氮、磷、硫等原子的共价作用来锚定单原子[35]。
碳元素在自然界中分布较为广泛,其多样化的存在方式决定了其多元化的功能[36]。将各类杂原子(如氮、磷)引入炭材料中可打破炭材料的电中性,改善炭材料的性能,从而对金属-碳(M-C)键的形成起促进和稳定作用,将金属单原子锚定在炭材料上[37]。而且炭材料本身兼具高比表面积、导电性好、耐腐蚀性和易改性等优点,是电催化的理想载体。因此,本综述着重介绍炭材料作为载体的单原子催化剂。
炭材料锚定金属单原子(M)的作用力也可分为两类:一类是在各类炭材料上引入杂原子如氮(N)以锚定金属单原子形成M-N-C 或N-M-C 结构作为活性中心;另一类是由于炭材料自身的独特结构具有能容纳金属单原子的缺陷,并能与金属单原子形成强的M-C 键作为活性中心。
1.1 炭载体上杂原子配位单原子催化剂
碳元素的异构体具有丰富的尺寸和微观结构[38],而碳纳米材料根据微观结构的不同可以被分为零维、一维、二维、三维,每种维度都有其特性[39]。其中,零维材料如碳量子点、富勒烯等具有量子尺寸效应﹑小尺寸效应、表面效应、宏观量子效应等;一维材料如炭纤维、碳纳米管、碳纳米线等,具有良好的电化学性能和力学性能,同时还有良好的吸附性能和较大的吸附容量;以石墨烯为代表的二维材料在力学、电学、热学和磁学等方面均显示出独特性能;三维材料也称块体材料,包括各种单一或复合的体相结构,除了同样具备电导性好、比表面积大等特点,由三维网络形成的大孔结构使多孔炭材料具有优异的吸附性能且质量轻、成本低。本部分内容着重介绍电催化二氧化碳还原的炭载催化材料中,利用杂原子实现金属活性组分的原子级分散的研究工作,并根据碳纳米材料维度进行分类。
1.1.1 一维炭载体
近年来,随着纳米材料研究的深入,一维碳纳米材料的应用前景不断被展现出来。一维材料中的碳纳米管、碳纳米棒具有许多优异的力学、电学和化学性能,而且重量轻,特殊结构连接完美,其中,碳纳米管的应用最广泛。碳纳米管(Carbon Nano-Tube,CNT)又名巴基管,其径向尺寸为纳米量级,轴向尺寸为微米量级,管子两端基本上都封口,主要由呈六边形排列的碳原子构成数层到数十层的同轴圆管。层与层之间保持固定的距离,约0.34 nm,直径一般为2-20 nm。
中国科学院大连化学物理研究所的黄延强研究员团队与新加坡南洋理工大学的刘彬教授[32]合作通过原位同步辐射和原位拉曼实验,阐述了碳纳米管负载单原子催化剂上ECR 反应的催化机制,揭示了Ni+是反应的活性位点。此外,通过动力学研究发现,Ni+能够高效活化CO2分子,而质子转移*C+H+→*COOH是反应的决速步骤。如图1 所示,他们构建了具有特定NiN4结构的四氨基酞菁镍分子,并通过重氮反应生成自由基而将其键连到碳纳米管上;球差电镜、X 射线吸收光谱证明了Ni 以单原子分散的形式存在,紫外光电子能谱发现CNT 上的部分电子会转移至NiN4活性位上。ECR 实验发现该材料可以高选择性转化CO2到CO,其法拉第效率可达99%,并且转化频率(Turnover frequency,TOF)高达10179 h-1。

图1 Ni SACs 模型及结构表征[32]:(a)CO2RR 模型Ni SACs 的合成;(b)Ni-CNT-CC 的FESEM 照片(比例尺,200 nm);(c)Ni-CNT-CC 的HAADF-STEM 照片(比例尺,5 nm);(d)Ni K-edge XANES;插图显示了EXAFS 光谱的傅里叶变换;(e)N K 边X 射线吸收光谱,其中峰A、B、C 和D 代表Ni-TAPc 中吡啶氮和桥接氮的1s–π*跃迁;(f)Ni-CNT-CC 和Ni-TAPc 的紫外光发射光谱的第二电子截止区;插入图显示了相同光谱的更宽区域(hv=40 eV,功函数(WF)=hv(40 eV)-Ecut-off)Figure 1 Structural characterization and the model of Ni SACs[32]: (a) Synthesis of model Ni SACs for the CO2RR,(b) FESEM image of Ni-CNT-CC (scale bar,200 nm),(c) HAADF-STEM image of Ni-CNT-CC (scale bar,5 nm),(d) Ni K-edge XANES;the inset displays a Fourier transform of the EXAFS spectra,(e) N K-edge X-ray absorption spectra,where peaks A,B,C,and D represent the 1s–π* transitions of pyrrolic and bridging aza nitrogen species in Ni-TAPc,(f) Second electron cut-off region of the ultraviolet photoemission spectra of Ni-CNT-CC and Ni-TAPc;the inset shows a wider region of the same spectra (hv=40 eV,work function (WF)=hv (40 eV)-Ecut-off)(with permission from Angewandte Chemie International Edition)
吴诗德等[40]以双氰胺为碳源和氮源,以乙酰丙酮镍为金属源,以氯化铵(NH4Cl)为第二氮源和造孔剂,采用NH4Cl 辅助热解-酸刻蚀的方法制备得到镍-氮-碳纳米管(Ni-N-CNT)ECR 催化剂,并考察了NH4Cl 添加量对催化剂结构和催化性能的影响。结果发现,当前体中加入NH4Cl 的量与氮源和金属源的总质量之比为1∶1 时,所得催化剂的催化性能最好。为了更好地解决金属原子团聚而导致的活性位点密度降低的问题,Pan 等[41]采用SiO2保护的煅烧策略合成由氮和铁共掺杂的碳纳米管结构(NFe-CNT,图2(a))。CNT 的层次结构提供了丰富的氮诱导缺陷,所以NFe-CNT 在ECR过程中也为电荷转移提供了一条畅通的途径,增强了协同效应,提高了ECR 催化活性。在低过电位0.48 V 时,Tafel 斜率为89 mV/dec,增强了CO在水溶液中的稳定性。同时,NFe-CNT 具有较大的界面面积和低阻扩散通道,更有利于提高活性位点的利用效率和促进物质迁移。这些研究旨在通过增加金属浓度或使用大比表面积炭载体(碳纳米管)的方法,提高M-Nx活性位点的暴露度,促进催化活性的提升。

图2 (a)SiO2 包覆焙烧工艺合成NFe-CNT 示意图[41],(b)Fe-N-C 催化剂合成过程[42]Figure 2 (a) Synthetic process for NFe-CNT/CNS by a SiO2-coated calcination strategy[41],(b) synthesis procedure of the Fe-N-C catalyst[42](with permission from Electrochimica Acta)
Wu 等[42]通过高温热解海胆状的FeOOH-PANI复合材料前驱体,制备了原子分散且高度暴露的铁氮配位活性中心掺杂的碳纳米棒状催化剂,如图2(b)所示。Fe-N-C-0.5 催化剂具有最佳的催化性能,在530 mV 的过电位和1.9 mA/cm2的电流密度下,对CO 具有高达95%的法拉第效率。催化剂具有高度暴露的Fe-Nx活性位点的多孔结构,以及较大的比表面积和电化学活性表面积,从而表现出优异的电催化活性。可见,提高M-Nx活性位点的密度和暴露度,促进传质,是提升ECR 催化剂活性的有效途径。
调控碳纳米管中不同类型的氮构型也是提高ECR 催化性能的方法。赵润瑶等[43]合成了以碳纳米管为载体的一系列铜单原子催化剂,由X 射线吸收谱分析表明,催化剂中的单原子铜位点分别与吡啶氮和吡咯氮配位。电催化性能测试表明,所制备催化剂可用于ECR 生成CO,由吡啶氮配位的Cu 单原子催化剂的反应选择性较差,而由吡咯氮配位的铜单原子催化剂其CO 法拉第效率在-0.70V(vs.RHE)时可达到96.3%,具有更高的活性。进一步研究发现,吡咯氮配位的铜单原子中心对于析氢反应具有更好的抑制效果。
1.1.2 二维炭载体
二维碳纳米材料以石墨烯(Graphene)为代表,是由sp2和sp杂化碳原子组成的新型碳同素异形体。早在2004 年合成以来,在电学、光学、热学等多个领域均已表现出优良的性能。近年来,由于石墨烯在电化学环境中的稳定性好、易形成比表面积大的层次孔结构、以及高导电性和高活性等优点,被广泛应用于电催化还原二氧化碳领域。
Zhang 等[44]提出了将石墨烯基体中嵌入CoN4活性位点,高度分散的CoN4位点表现出良好的ECR 活性,CO 的法拉第效率在-0.76 V(vs.RHE)下接近95%,对应的过电位为0.65 V,且具有优异的稳定性。刘灵惠[45]针对石墨烯基单原子催化剂在高温合成中碳层堆叠、金属聚集问题,采用自制蒙脱石为模板,制备了石墨烯纳米片层负载过渡金属单原子催化剂。实验结果表明,这种特殊的结构使得Ni-SAC 催化剂在ECR 中具有优异的催化活性,在(-0.6)-(-1.1) V(vs.RHE)的大电位,CO 法拉第效率大于90%,并且能在连续30 h 的恒电位测试(-0.9 V vs.RHE)中保持稳定以及CO 法拉第效率维持在95%。另外石墨烯上原子分散的氮配位铁位点(Fe-N4)也能够高效催化CO2转化为CO。Pan 等[46]报道了通过对石墨烯边缘孔隙的调控来改变孤立Fe-N4位点的局部电子结构可显著提高ECR 过程,其CO 法拉第效率为94%,在0.58 V(vs.RHE)下的TOF 为1630 h-1,明显优于孔隙稀缺的Fe-N-G(是前者的三倍)。与石墨烯负载的Fe-N4相比,这种电子修饰通过增加CO 吸附构型中Fe-C 键的长度和强度,减弱了*CO 中间体在孔载Fe-N4位点上的吸附,从而促进了*CO 的解吸过程。图3(a)所示为孔隙稀缺和孔隙丰富的石墨烯负载Fe-N4的ECR 对比示意图。
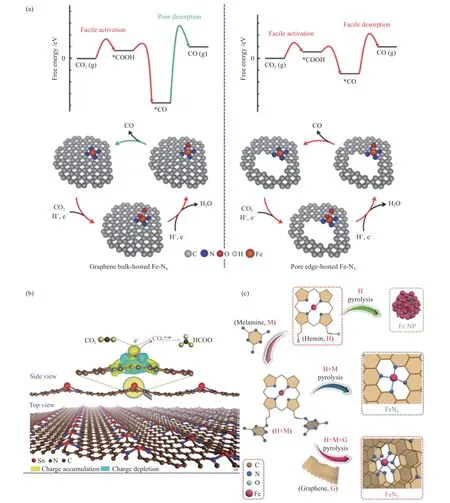
图3 (a)左图为孔隙稀缺的石墨烯负载Fe-N4;右图为孔隙丰富的石墨烯负载Fe-N4[46];(b)为氮掺杂石墨烯上单原子Snδ + 的三维差分电荷密度图和CO2 电还原成甲酸盐示意图(黄色和蓝色等值面分别对应电子数量的增加和耗竭区)[47];(c)为单原子FeN4 和FeN5 催化剂的合成路线[49]Figure 3 (a) Schematic showing the electrocatalytic CO2 reduction behaviors on pore-deficient graphene bulk-supported Fe-N4 (the figure on the left) and pore-rich graphene-supported Fe-N4(the figure on the right)[46],(b) the figure is plots of 3D differential charge densities of the single-atom Snδ + on N-doped graphene and schematic illustration for CO2 electroreduction into formate (the yellow and blue isosurfaces correspond to the increaseinthe number ofelectronsand thedepletionzone,respectively)[47],(c) synthetic route towards single-atomFeN4and FeN5catalysts[49](with permission from ACS publications,Advanced Materials,and Angewandte Chemie International Edition)
此外,通过改善微观价键结构来调节中间体,调控反应方向,对于ECR 产物的选择性有着决定性的作用。谢毅院士团队[47]发现,将Sn 以原子状态分散在石墨烯表面会使Sn 带正电荷,有利于CO2的活化与质子化,增强甲酸盐的选择性,如图3(b)所示。并且键长相对较短的Sn-N 键使得催化剂能够保持较高的化学稳定性,在200 h 的连续电解中,ECR 法拉第效率及电流密度基本没有下降[48]。Zhang 等[49]通过在石墨烯表面的自组装和碳化处理得到的N 掺杂石墨烯锚定的单原子Fe 催化剂,如图3(c)所示,在热解过程中FeN4位点与N 掺杂石墨烯的吡咯N 相互作用形成FeN5构型。密度泛函理论(Density Functional Theory,DFT)计算表明,吡咯N 配位可有效抵消Fe 3d轨道的电子密度及降低Fe-CO 的 π反馈效应,有利于CO 的快速解吸和高选择性。
1.1.3 三维炭载体
具有多孔结构的炭材料,不仅具有成本低、比表面积大、电导率高、化学稳定性好等优点,且其高度发达并可调控的孔结构使其拥有众多的边缘位置及丰富的活性位点,十分有利于电子传输[50],故在电催化材料领域中脱颖而出。在炭基材料中加入杂原子可以有效地修饰催化剂的电子结构和催化性能,杂原子氮的孤对电子可以捕获金属中心[51],形成稳定的金属位点[52,53],是掺杂于多孔炭的理想元素。氮掺杂多孔炭材料(Nitrogen-doped Porous Carbon materials,NPC)作为电荷存储和电子传输载体,在催化领域有重要应用,已成为炭材料领域的研究热点之一[54]。
Varela 等[55]制备了含Fe、Mn 的M-N-C 单原子催化剂对ECR 进行了研究,还原过程中选择性及作用机制如图4(a)、(b)所示。结果显示氮掺杂多孔炭载体单原子催化剂,一方面,在ECR 产生CO/H2混合物中具有高活性和高CO 选择性;另一方面,通过增强Fe 的活性中心以及其与CO 的相互作用能够将CO 氢化成碳氢化合物。该实验证明了M-N-C 材料对 ECR 具有广阔的应用前景,并有望替代在水相电解质中还原CO2的贵金属(Au、Ag)催化剂。
氮掺杂多孔炭负载的金属单原子催化剂有多种制备方法[56],常见的有炭载体掺杂法、混合碳化法、有机骨架前驱体法、氮掺杂炭载体上金属配合物锚定法、热解法等。而研究者们更关注的是如何降低NPC 负载的金属单原子催化剂的制备成本。Hu 等[57]利用混合碳化法制得Fe、Ni、Co 单原子催化剂。混合碳化法是利用廉价的邻苯二胺作为含氮前驱体、二氧化硅纳米颗粒作为模板与三种过渡金属(Fe、Co、Ni)氯化物中的任何一种发生热解从而形成含有二氧化硅和金属颗粒的炭材料。将得到碳化产物经NaOH 溶液蚀刻和H2SO4浸洗后进行二次热解最终得到含有MN4结构的M-N-C 材料,如图4(c)所示。通过分别考察它们对ECR 的活性发现Ni 掺杂的多孔炭选择性最高,CO 的法拉第效率为93%。Ye 等[58]采用富氧金属-有机骨架(Zn-MOF-74)前驱体,通过优化焙烧温度和时间调节活性氮的含量,制备了具有大量的活性氮(吡啶和石墨性氮)和多孔结构的NPC,表现出优异的ECR 活性,具有-0.35 V 的低起始电位和高的CO 法拉第效率,在-0.55 V(vs.RHE)时达到98.4%,为在实际应用中制备高活性、低成本的贵金属ECR 催化剂提供了一种有效且简便的方法。
金属单原子本身与载体的配位情况也会影响M-N-C 的催化效果。Peter Strasser 课题组[59]以混合热解合成了Ni-N-C 和Fe-N-C 催化剂,与合成的无金属N-C 作对照,如图4(d)。并且成功地将无贵金属的固体Ni-N-C 电催化剂应用于微流池电解槽的气体扩散电极中将CO2还原至CO。在环境压力和温度以及阳极液和阴极液的中性pH 值下,验证了 Ni-N-C 催化剂在50-700 mA/cm2的工业电流密度下比商业Ag/AgOx催化剂、Fe-N-C 以及无金属氮掺杂催化剂具有显著的稳定性和效率优势。
Zhao 等[60]先通过水热法合成了掺杂Cu 的ZIF-8然后在1000 ℃的N2气氛下对该前驱体进行碳化反应制得了Cu-SA/NPC,并指出Cu 与四个吡咯氮原子的配位结构是主要的活性位点,能够降低CO2活化能和C-C 偶联所需的反应自由能。由图4(e)可见单原子Cu 加入NPC 材料可以显著提高其CO2还原催化活性。值得注意的是,该反应能在较低的-0.16 V 电位下产生氧合产物,见图4(f)。其中,CH3COCH3是Cu-SA/NPC 催化剂在低电位下ECR 的主要产物。
硫与氮类似,也是p区元素,冯丽芳[61]通过简单的固相热解法合成了一种将Ni 锚定在硫、氮共修饰的多孔炭(Ni SAs/NS-C)上的单原子催化剂,其活性中心为NiN4-S1(硫和氮相连)。该催化剂在低电位下表现出相对较高的 ECR 活性与稳定性,实验与DFT 计算结果表明,引入硫可对Ni-N4结构中原有的高度对称的电子结构产生影响从而使Ni SAs 表面呈现富电子状态,降低了其对中间产物的反应活化自由能。但同时由于硫的原子半径与氮差异较大,可能会在氮掺杂多孔炭上形成缺陷进而产生“电子陷阱”[62],因此,如对多种杂原子共掺杂的电子结构还有待进一步研究。ACHAADF-STEM 和元素分析测试显示了Ni、C、N、S 的元素分布,如图5(a)所示,可以发现各种元素是均匀分布的,也验证了Ni、N 和S 元素的成功掺杂。
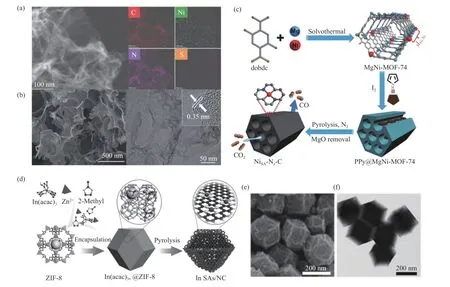
图5 (a)Ni SAs/N-C 的Ni SAs/NS-C 电催化剂中C、N、Ni、S 元素的分布[61];(b)NPCA900 的SEM 照片(左)和NPCA900 的TEM 和HR-TEM 照片(右)[62];(c)用于电催化CO2 还原的NiSA-Nx-C 催化剂制造的主-客体合作保护策略[65];(d)In-SA/NC 的制备流程示意图;(e)In-SA/NC 的SEM 照片;(f)In-SA/NC 的TEM 照片[66]Figure 5 (a) Distribution of C,N,NI and S elements in Ni SAs/NS-C electrocatalysts of Ni SAs/N-C[61],(b)SEM images of NPCA900(the figure of left),TEM and HR-TEM images of NPCA900 (the figure of right)[62],(c) Illustration showing the host-guest cooperative protection strategy for the fabrication of NiSA-Nx-C catalysts for electrocatalytic CO2 reduction[65],(d) Schematic illustration for the preparation of In-SAs/NC,(e) SEM,(f) TEM[66](with permission from Angewandte Chemie International Edition)
炭气凝胶(Carbon Aerogel,CA)是一种轻质、多孔、非晶态的纳米炭材料,其连续的三维网络结构可在纳米尺度进行调控,因具有孔隙率高、比表面积大、电导率高(为25 S/cm)等优点,所以是理想的电极材料[63]。Chen 等[62]使用氮、磷共掺杂炭气凝胶来增强ECR 到CO 的过程,并将其命名为NPCA900,同时用类似方法合成了氮掺杂炭气凝胶(NCA900),磷掺杂炭气凝胶(PCA900)和炭气凝胶(CA900)作对照。从SEM 和TEM 可以观察到,所制备的NPCA900 具有多孔网络状结构,由类石墨烯柔性超薄纳米片组成,如图5(b)所示。氮、磷共掺杂炭气凝胶比氮掺杂或磷掺杂炭气凝胶具有更高的电化学活性面积和电子电导率,有利于CO2的电子转移,CO 的FE 可以达到99.1%,部分电流密度为-143.6 mA/cm2。
金属有机骨架化合物(Metal Organic Frameworks,MOFs)是由无机金属中心(金属离子或金属簇)与桥连的有机配体通过自组装相互连接,形成的一类具有周期性网络结构的晶态多孔材料[64]。Gong 等[65]开发了一种通用的主客体合作保护策略来构建SAC。如图5(c)所示,在MgNiMOF-74中引入Mg2+延长了相邻Ni 原子之间的距离;在热解过程中将聚吡咯(PPy)客体作为N 源引入双金属-有机框架中来稳定分离的Ni 原子,并通过控制热解温度制备了一系列具有不同N 配位数的单原子Ni 催化剂(NiSA-Nx-C)。理论计算表明,NiSANx-C 中单原子Ni 位点的低N 配位数更有利于COOH*中间体的形成,NiSA-N2-C 表现出高的CO 法拉第效率(98%)和TOF(1622 h-1)。
Shang 等[66]设计了一种新的铟(In)单原子催化剂,该催化剂在金属有机框架(MOFs)衍生的N 掺杂碳基质(InSAs/NC)上具有独特的 Inδ+-N4原子界面部分,可以作为一种高效的ECR 至HCOO-的电催化剂。In-SAs/NC 的制备工艺包括湿法浸渍工艺和热解工艺,其制备流程示意图如图5(d)所示,并通过SEM 和TEM 对In-SAs/NC 的形貌进行了表征,如图5(e)、(f)所示,呈规则多面体结构。测试其电催化性能,发现InSAs/NC 在-0.95 V(vs.RHE)下的TOF 为12500 h-1,在-0.65 V(vs.RHE)下,ECR 至HCOO-的最大法拉第效率为96%。
1.2 炭载体上碳原子配位单原子催化剂
除了需要通过杂原子桥联实现金属与炭载体的锚定,还可通过强的金属-碳键稳定金属单原子活性中心,进而实现二氧化碳电还原。二维金属碳化物(Mxene)和石墨炔便是两种典型的不用引入其他杂原子,依靠金属-碳键的强作用力将金属单原子稳定锚定的两种炭材料。
1.2.1 二维金属碳化物
Mxene 材料是一类具有独特二维层状结构的金属碳化物。Mxene 是通过对 MAX 相(由强金属、离子和共价键连接)中的 A 元素进行选择性蚀刻而产生的具有超高导电性和丰富表面官能团的一类新材料[67]。Zhang 等[68]发现,Ti 锚定的单层Ti2CO2(一种Mxene 材料)对CO 的氧化过程有极高的催化活性。
Zhao 等[69]在室温下利用超薄的二维Ti3–xC2Ty型 Mxene 纳米片通过自还原方法制备稳定化的Pt 单原子催化剂。Ti3–xC2Ty的每一层由三种Ti 子层组成,碳位于八面体的间隙位置,相邻两层之间由活性更强的Al 层连接。他们发现,在用氢氟酸处理并剥离单层Mxene 时,一些邻近的Ti 原子也会被蚀刻掉,产生了丰富的Ti 空缺位点可以用来容纳金属单原子。此外,被剥落的Mxene 片具有强还原性,也有利于稳定单原子与其形成的强金属-碳键。他们进一步研究在Pt1/Ti3–xC2Ty和还原剂三乙基氢硅烷的存在下CO2(~ 1 atm)与苯胺生成甲酰胺的反应,发现该反应实现了近乎100%的转化率和选择性,见图6(a)。
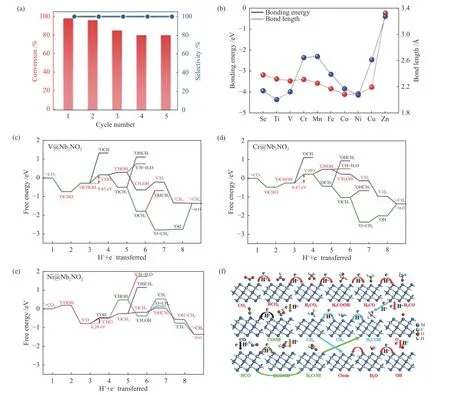
图6 (a)为Pt1/Ti3-xC2Ty 催化苯胺N-甲酰化反应的回收试验[69];(b)第四周期过渡金属与Mo2CS2 之间的能带以及和硫原子之间的键长[70];(c)、(d)、(e)为在U=0 V 下,TM@Nb2NO2 上ECR 产生CH4 的吉布斯自由能图,其中,红色路线为最有利反应途径[71];(f)为以化学式M3C2 作高效催化剂在MXenes 上发生ECR 得到不同产物的可能机理途径,其中,红色路线为最有利反应途径[73]Figure 6 (a) Recycling test of Pt1/Ti3–xC2Ty for the catalytic N-formylation of aniline[69],(b) Banding energy between TMs of the fourth period and Mo2CS2 and the bond length between TM and S atoms[70],(c),(d),(e) Gibbs free energy diagram for ECR on TM@Nb2NO2 to produce the final product CH4 under U=0 V;the red pathway denotes the optimal pathway[71],(f) Proposed ECR possible mechanism pathways to different products on MXenes with the formula M3C2 as high efficiency catalysts;the red pathway denotes the optimal pathway[73](with permission from ACS publications,Chemistry -A European Journal and Elsevier)
由于非贵金属不仅成本低廉而且储量丰富,研究者们更愿意用非贵金属替代贵金属制备催化剂。过渡金属(Transition Metal,TM)负载的Mxene催化剂对ECR 的选择性均高于HER,因此,研究过渡金属碳化物催化剂是非常有意义的。Li 等[70]通过明确界定的密度泛函理论(DFT)计算研究了Mo2CS2型MXene 的第四周期过渡金属单原子催化剂对ECR 的催化性能。他们先制备出类似固氮酶结构的新型Fe@Mo2CS2,发现其能激活对CO2转化几乎不起作用的Mo2CS2。在此基础上进一步研究得出,除Cu 外的九种TM@Mo2CS2非常稳定,见图6(b),除Cu 和Ni 外的八种TM@Mo2CS2可有效抑制HER,而Cr、Fe、Co、Ni@ Mo2CS2对ECR都表现出良好的选择性和催化活性。
CO2加氢制甲烷(CH4)是CO2还原的主要途径之一,这种转化方式既实现了碳循环利用,又合成了新的能源,因此引起了广泛关注。Lu 等[71]通过对Nb2NO2型Mxene 材料进行DFT 计算,得出V、Cr、Mn、Fe、Co、Ni 原子能稳定地嵌入氮的空缺位点且不会发生聚集。在所有反应可能产生的C1产物中,CH4的气体吸附能最小,因此,该ECR 过程最有可能得到CH4。不同的过渡金属碳化物参与反应的电位决定步骤不同,且V/Cr/Ni@Nb2NO2对ECR 得到CH4过程表现出高催化性能,见图6((c)-(e)),其较小的限制电位分别为-0.45、-0.47和-0.28 V,优于Cu(211)的-0.74 V。同样是对V、Cr、Mn、Fe、Co、Ni 这六种过渡金属单原子催化剂进行理论研究,Li 等[72]利用T=-O 和T=-S 两种官能团构建了Ti2C 基上负载这六种过渡金属的单原子催化剂,通过调控MXene 表面官能团可调控MXene 基催化剂的二氧化碳电催化性能。他们对比发现,当ECR 生成CH4时,TM@Ti2CS2不仅能活化CO2分子,还能降低反应能垒,TM@Ti2CS2的催化性能优于TM@Ti2CO2。针对产物为CH4的ECR 过程,Xiao 等[73]扩大了研究范围,研究了M3C2型MXenes(M=Sc、V、Cr、Mn、Zr、Nb、Mo、Mo2Ti、Hf、Ta、W)的催化性能。研究发现,过渡金属不同的活性位点使CO2沿着不同路径转化产生不同的烃类产物,其中,Ti3C2、Ta3C2和Sc3C2能最大限度地将CO2还原为CH4。当反应沿着生成CH4的最有利途径进行时,见图6(f)中的蓝线,其限制电位为-2.10 和-0.89 eV,分别低于大多数Ta3C2和W3C2生成H2O 的限制电位-1.43 和-0.74 eV。Li 等[72]还研究了ECR 生成COOH/HCOO 的过程,这时TM@Ti2CO2对CO2分子的活化程度更优。当CO2分子吸附在TM@Ti2CO2上时会发生轨道重构从而发生形变,且能提供额外的电子利于CO2的活化,因此锚定的金属单原子不同活化效果也有所差异。
1.2.2 石墨炔
石墨炔(Graphdyne,GY)是一种二维炭材料,含有丰富的C≡C 键和分布均匀的孔隙,具有较大的共轭结构和“炔-烯互变”性质。石墨炔结构中的sp杂化碳原子能与金属原子形成强的金属-碳共价键,从而使金属原子能被稳定地锚定在其上,是一种理想的单原子催化剂载体。
Ni 等[74]研究了不同孔径石墨单炔(Graphyne,GY)的骨架结构对锚定的Cu 单原子的影响,发现石墨炔的孔径大小会影响Cu-GY 的活性,如图7(a)所示。这是因为如果石墨炔的孔径改变,Cu 原子的配位环境和石墨炔对中间体的空间位阻会同时改变,进而会导致Cu-GY 的ECR 催化性能改变。相比之下,具有较大孔隙的石墨炔因空间位阻较小从而对ECR 的催化性能最好。Shi 等[75]通过将Cu 单原子在石墨炔上的原位锚定得到Cu SAs/GDY,构建的Cu-C 键可以提供高效的电荷转移通道,控制反应中间体,促使生成CH4的反应更容易进行,从而显著提高其催化性能,图7(c)为合成示意图。
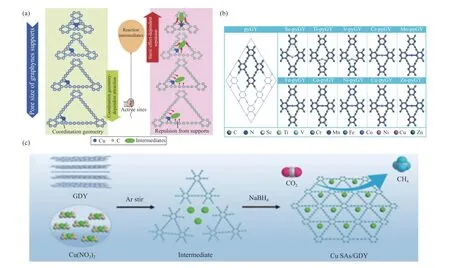
图7 (a)为石墨炔负载Cu 单原子ECR 活性的两个影响因素[74];(b)为pyGY 和TM-pyGYs 的结构[76];(c)为Cu SAs/GDY 的形成示意图[75]Figure 7 (a) Two influencing factors of ECR of graphyne-supported Cu single atoms[74],(b) The structures of pyGY and TM-pyGYs[76],(c) Schematic illustration of the formation of Cu SAs/GDY[75](with permission from Angewandte Chemie International Edition and RSC Publications)
Wang 等[76]对第一排过渡金属嵌入吡嗪基石墨炔(TM-pyGYs)作为ECR 的潜在SAC 进行了研究,见图7(b),计算得出金属原子与pyGY 之间存在较强的共价作用、较大的电荷转移和明显的轨道重叠,因而TM-pyGYs 在ECR 过程中具有良好的稳定性。研究发现,大多数TM-pyGys(除Sc/ZnpyGY)都能抑制HER,其中,HCOOH 是Ti/Cr/Mn/Fe/Ni/Cu-pyGygs的主要产物,CH4是V/Co-pyGYs的主要产物。他们还发现,外加电位对TM-pyGY的ECR 活性和产物选择性有显著影响。在没有外加电位时倾向于在Ti/Cr/Mn/Fe/Ni/Cu-pyGygs 上产生HCOOH;当施加的电位超过-0.4 V 时,Fe/Co/NipyGY 具备能产生高通量CH4的优点。
Pan 等[41]在石墨二炔上利用乙炔键的导向作用捕获位点合成了从原子级到亚纳米级团簇再到纳米团簇的一系列可控尺寸的Cu 催化剂,为研究不同尺寸的过渡金属催化剂提供了一条可行的思路。
2 总结与展望
炭材料负载的单原子催化剂在电还原二氧化碳领域表现出独特的性能,根据活性中心结构的不同,本工作重点介绍了通过杂原子桥联形成金属-杂原子-碳和金属-碳键两种方式构筑单原子催化活性中心的炭载催化剂,总结了这些炭基材料负载的单原子催化剂的制备、结构、性能及在ECR领域的应用。对于还原产物的组成,由于CO2还原后生成的CO 从活性位点脱附后较难扩散到另一个单原子位点,碳-碳偶联发生的可能性较低,目前,单原子催化剂上主要为C1小分子产物为主,例如CO 和CH4。
在接下来的研究中,炭载体的设计、与金属中心的相互作用、反应机理、原位表征等方面仍有深入的空间。对于载体的结构,可优化载体的结构改变碳原子的局域电子密度,从而更有利于反应物、中间体的吸附活化和产物脱附,提升催化剂活性。目前,对于ECR 活性中心的具体结构和反应中的作用机制尚存争议,载体表面缺陷位种类数量及金属-载体的构型,均会影响反应进程,从而决定最终产物的选择性,如果能够构筑具有特定结构的催化剂活性中心,将有利于确定活性中心与反应历程的构效关系。在此过程中,还需要借助原位表征手段,跟踪反应过程中催化剂的结构演化及反应中间物种的识别,为实际反应过程的机理研究提供直接证据。
