含脱水剂的碳酸二甲酯直接合成法催化体系综述
2023-11-21程庆彦谷云含王锦涛王延吉乔金栋
程庆彦,张 帅,谷云含,王 茁,王锦涛,李 莉,王延吉,王 寰,乔金栋
(1.河北工业大学 河北省绿色化工与高效节能重点实验室 天津市本质安全化工技术重点实验室,天津 300401;2.海驰创研(天津)科技有限公司,天津 300392;3.天津杰斯曼建筑材料有限公司,天津 300382)
自工业革命以来,人类向大气中排放的CO2等温室气体逐年增加,其引发的一系列问题已引起世界各国的关注[1]。有效控制和减少CO2排放成为所有国家的紧迫任务[2]。CO2作为一种丰富、无毒且廉价的碳源,将其转化为燃料或与燃料相关的产品[3]可以解决上述问题,如利用CO2合成甲醇、甲酸盐、尿素、碳酸二甲酯(DMC)等。常温下,DMC 是一种无色透明的液体,属低毒或无毒类化学品[4]。DMC 分子中含有羰基、甲基和甲氧基等官能团,可以替代光气用作甲基化试剂,也可以替代剧毒且致癌的硫酸二甲酯(DMS)用于农药、医药中间体及抗氧剂、塑料加工稳定剂等重要的精细化工产品生产中[5]。DMC含氧量高达 53%,可代替甲基叔丁基醚(MTBE)用作柴油添加剂,提高汽油辛烷值,降低环境污染,减少CO 和NOx排放[6]。DMC 已被用作锂离子电池的电解质,并用作非质子极性溶剂[7]。DMC 还具有使用安全、污染少、运输方便等优点[8]。
DMC 合成方法主要有光气法、甲醇氧化羰基化法、酯交换法、尿素醇解法、CO2与CH3OH 直接合成法[9-11],这些合成方法及优缺点归纳于表1 中。
CO2和CH3OH 直接合成DMC 使用廉价易得的原料,反应原子经济,副产物少,是一条绿色合成路线,已成为近年来的研究热点。由于该反应的热力学平衡限制及CO2和CH3OH 的活化问题[12],催化剂的设计、应用、回收及高效脱水剂的选择尤为重要。
1 反应热力学
研究2CH3OH(l)+CO2(g)→DMC(l)+H2O(l) 反应的热力学数据主要包括反应标准摩尔焓变、反应标准摩尔熵变、标准摩尔吉布斯自由能变、摩尔定压热容和平衡常数[13-16]等,计算中所涉数据见表2。

表2 CO2 和CH3OH 合成DMC 反应中化合物的热力学数据和比热容系数(298.15 K,1.0 × 105 Pa)Table 2 Thermodynamic data and specific heat capacity coefficients of various compounds in the synthesis of DMC from CO2 and CH3OH at 298.15 K and 1.0×105 Pa
反应标准摩尔焓变:
化学反应的标准摩尔熵变:
标准摩尔吉布斯自由能变:
化学反应达平衡时,标准摩尔吉布斯自由能变与反应的标准平衡常数的关系:
在恒定压力下,反应摩尔焓变 ∆rHm和摩尔吉布斯自由能变 ∆rGm与温度的关系可分别表示为:
反应的摩尔定压热容变化值 ∆rCp,m的计算如下:
根据基团贡献法计算化合物CH3OH、CO2、DMC、H2O 的热容值[17],以二阶多项式形式表示化合物中不同基团对温度的依赖性:
式 中,AC、BC、DC为CH3OH、CO2、DMC、H2O的比热容系数。
经计算,反应摩尔定压热容变化值与温度的关系式为:
式中,Ar=-20.1828,Br=19.294×10-2,Dr=-4.9631×10-4。
结合式(5)、(6)、(8)、(9)可得反应摩尔焓变∆rHm和 摩尔吉布斯自由能变∆rGm与温度的关系分别为:
将Ar、Br、Dr、R、∆r、∆r、α=15187.25、β=-910.4代入式(10)和(11),得到:
由(4)、(11)可得平衡常数与温度的关系为:
甲醇平衡转化率可表示为:
反应摩尔吉布斯自由能变与温度和压力的关系式可表示为:
将式(13)、p0=1.0×105Pa、∆n=-1(该反应中生成物和反应物气体摩尔数之差),代入上式,可得:
由式(1)、(3)、(12)可知,在标况下,该反应为非自发放热反应,热量释放较少[18-20],随着温度升高释放的热量增加;由式(13)、(14)计算可知,标况下反应平衡常数Ka=6.137×10-5,随着温度升高,吉布斯自由能增大,平衡常数减小,不利于反应正向进行;由式(17)可知,当反应温度为453.15 K,二氧化碳初始压力超过1.61× 107Pa时,∆rGm<0;在实践中,在一定范围内降低反应温度和增加压力可提高该反应的平衡转化率。总之,基于热力学分析,在适宜反应条件下,由CO2和CH3OH 直接合成DMC 是可行的。
2 CO2 直接合成DMC 的催化剂研究
2.1 离子液体催化剂
离子液体(ILs)是指全部由离子组成的液体,被认为是相对清洁的催化剂,其可操作温度范围宽((-40)–300 ℃),具有良好的热稳定性和化学稳定性等优势。近年来,ILs 已用于催化CO2和CH3OH合成DMC。
2013 年,Sun 等[21]制备出一系列羟基功能化的碱性ILs,命名为氢氧化胆碱(CH)。在最优条件下,CH3OH 转化率为0.60%,DMC 选择性可达95.2%(表3)。CH 回收并重复使用五次后,催化活性几乎不变。
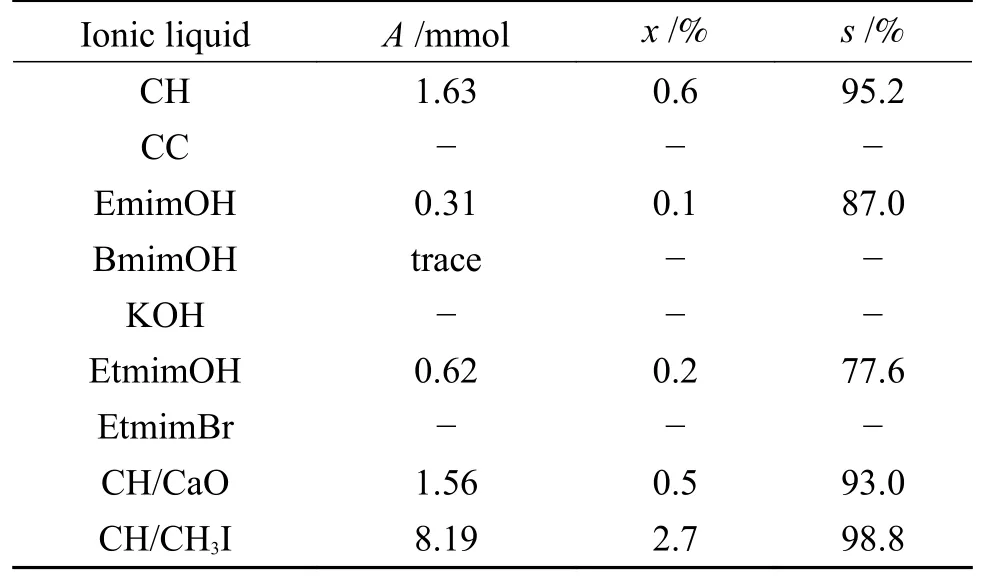
表3 离子液体催化剂上CO2 和CH3OH 合成DMC 催化性能[21]Table 3 Catalytic performance of various ionic liquids (ILs)for the synthesis of DMC from CO2 and CH3OH[21]
Zhao 等[22]制备出咪唑鎓碳酸氢盐离子液体催化剂[CnCmIm][HCO3](Cn和Cm表示碳链长度)。室温下CH3OH 转化率高达74.0%,当反应温度为50 ℃时,CH3OH 转化率为82%,DMC 选择性为94%(表4)。高CH3OH 转化率主要是由于离子液体[CnCmIm][HCO3]同时充当催化剂和脱水剂,其催化和脱水机理如图1 所示。在该离子液体系统中,CO2加合物(CnCmIm-CO2)和离子液体[CnCmIm][HCO3]之间存在平衡,CnCmIm-CO2可以促进CO2和CH3OH生成DMC 和H2O;同时CnCmIm-CO2也可以与H2O结合,推动平衡移动,促使DMC 形成。图2 为CO2和CH3OH 合成DMC 的能量分布计算,从[CnCmIm][HCO3]到CnCmIm-CO2转变表明,碱性HC能以很低的能垒(19.6 kcal/mol)与N-CH-N 中质子结合,在室温下就能进行;此外,CnCmIm-CO2与H2O 反应速率非常快,在20 min 内可以达到平衡,从CnCmIm-CO2到[CnCmIm][HCO3]的能垒仅为19.0 kcal/mol,这种低能垒使反应在常温下能够快速进行。富含电子的Lewis 碱[23]可以直接结合并活化CO2形成加合物,例如N-杂环卡宾[24]、脒基和胍基碱[25]、烷基磷[26];CnCmIm-CO2可以与CH3OH中质子结合,产生活性CH3O–阴离子,并以低势垒与CO2结合形成 CH3OC;另一CH3O–通过Int2再次与CH3OH 中质子结合;最后,通过Int3 脱水生成DMC 和中间体CnCm,CnCm可以结合CO2再次形成Rea,从而进入下一个循环。该反应首先在催化剂的Lewis 碱性位点上活化CO2,之后催化剂再与CH3OH 中质子结合形成活化态的CH3O–中间体,活化的CO2与CH3O–结合生成产物DMC,这一过程与先活化CH3OH 再活化CO2的机理不同[27]。

图1 [CnCmIm][HCO3]上CO2 和CH3OH 合成DMC 的催化和脱水机理[22]Figure 1 Catalytic and dehydration mechanism of synthesis of DMC from CO2 and CH3OH on the [CnCmIm][HCO3] catalyst[22](with permission from John Wiley and Sons)
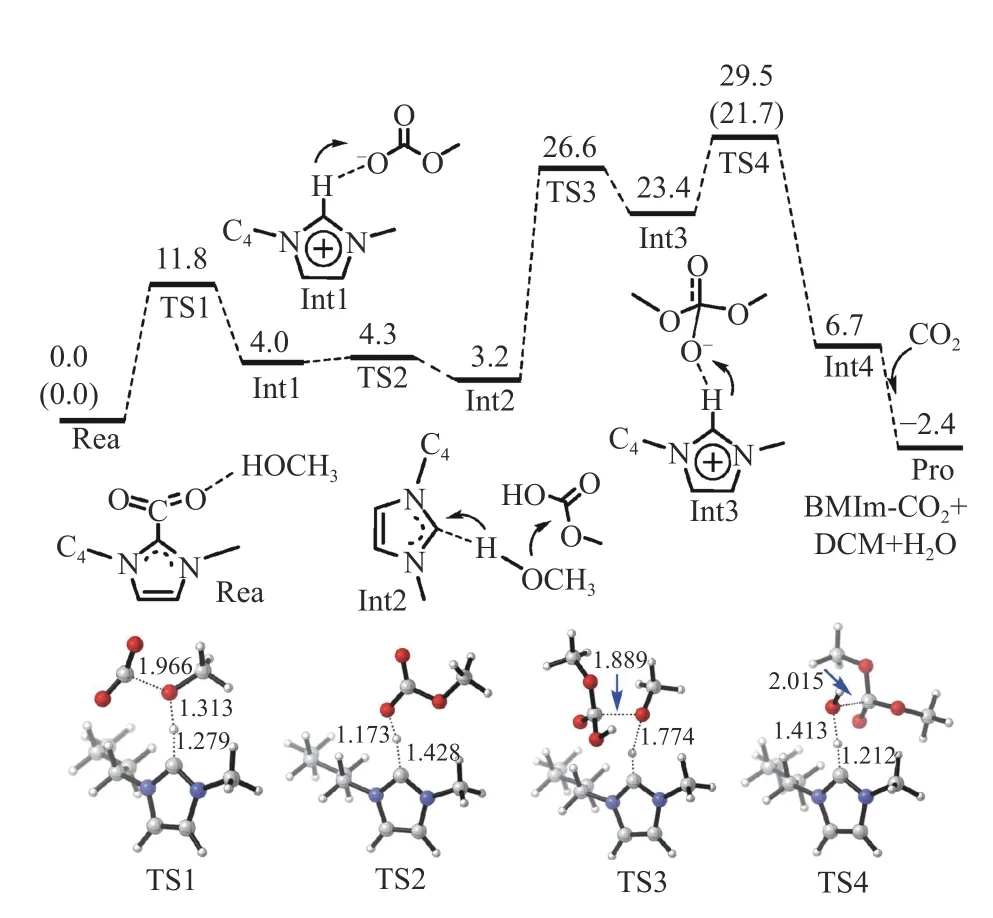
图2 CO2 和CH3OH 合成DMC 的能量分布(单位:kcal/mol),Int1-4 和TS1-4 为优化后的中间产物模型(键长单位Å)[22]Figure 2 Energy distribution for synthesis of DMC from CO2 and CH3OH (unit: kcal/mol),Int1-4 and TS1-4 are optimized intermediate models (bond length unit Å)[22](with permission from John Wiley and Sons)

表4 [CnCmIm][HCO3]催化剂上CO2 和CH3OH 合成DMC 催化性能[22]Table 4 Catalytic performance of [CnCmIm][HCO3] catalysts for the synthesis of DMC from CO2 and CH3OH[22]
ILs 催化剂通常是复杂的,至少由两种组分组成,绝大多数ILs 在单独使用时DMC 选择性超过90%,而CH3OH 转化率低于10%,一些具有脱水功能的ILs 可使CH3OH 转化率提高数十倍。
2.2 碱金属碳酸盐催化剂
2001 年,Fujita 等[28]考察各种碱金属碳酸盐催化CO2和CH3OH 合成DMC 性能,发现K2CO3是最有效的催化剂,主要副产物是二甲醚(DME),如表5 所示,在温度70 ℃和CO2压力8.0 × 106Pa下,在CH3I 参与下,CH3OH 转化率为4.1%,DMC选择性为81.8%。CH3I 辅助碱金属碳酸盐催化CO2和CH3OH 合成DMC 反应机理如图3 所示,首先,CH3OH 中质子与催化剂碱性位点结合,生成甲氧基阴离子CH3O-;CH3O-与CH3I 或CO2反应分别生成DME 或碳酸甲酯阴离子[CH3OCH2]-;随后,[CH3OCH2]-与CH3I 生成DMC,I-与催化剂碱性位点上质子发生反应,催化剂恢复原状,产物HI 可以和CH3OH 进一步反应生成CH3I。
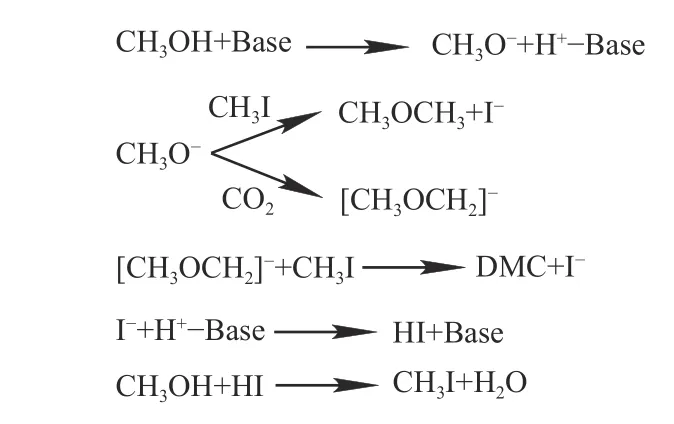
图3 CH3I 参与下CO2 和CH3OH 合成DMC 反应机理[28]Figure 3 Reaction mechanism of synthesis of DMC from CO2 and CH3OH with the participation of CH3I[28](with permission from ACS Publication)
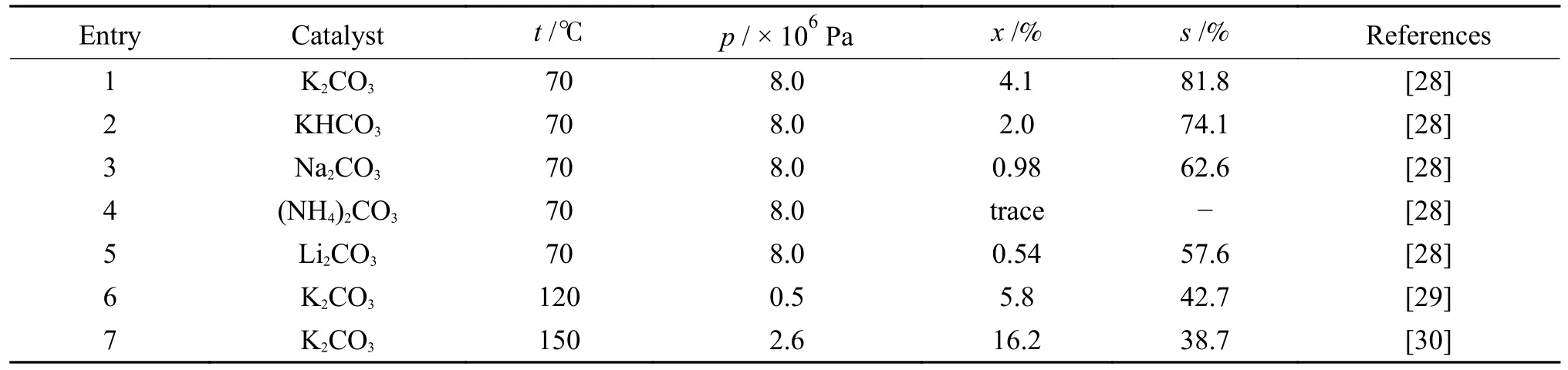
表5 碱金属碳酸盐催化剂上CO2 和CH3OH 合成DMC 催化性能Table 5 Catalytic performance of alkali metal carbonate catalysts for the synthesis of DMC from CO2 and CH3OH
Liu 等[29]开发出一种用于K2CO3催化CO2和CH3OH 合成DMC 的催化体系,在CO2压力5.0 ×105Pa,反应温度120 ℃和环氧乙烷存在下反应6 h,CH3OH 转化率为5.8%,DMC 选择性为42.7%,主要副产物是碳酸丙烯酯(PC)和1,2-丙二醇(表5)。
为进一步提高CH3OH 转化率,Yang 等[30]使用K2CO3作为催化剂,在氧化环己烯存在下由CO2和CH3OH 合成DMC。在反应温度150 ℃和CO2压力2.6 × 106Pa 下,CH3OH 转化率为16.2%,DMC 选择性为38.7%。尽管CH3OH 转化率从5.8%增加到16.2%,但CH3OH 转化率仍较低(表5)。
对于该反应,碱金属碳酸盐催化剂的主要优点是相对便宜和易获取,但这种催化过程需要昂贵的CH3I 辅助。通常,碱金属碳酸盐上DMC收率在1%–6%,比其他催化剂低。
2.3 过渡金属氧化物催化剂
过渡金属氧化物是CO2和CH3OH 合成DMC的一大类有效催化剂,因其活性组分是金属氧化物,不存在氧化失活的问题,可以在空气中储存很长时间,这对于工业应用具有很大优势;缺点是DMC收率低,需通过添加助催化剂来改善。表6 为在不同温度和压力下,金属氧化物上CO2和CH3OH合成DMC 的催化活性。

表6 过渡金属氧化物催化剂上CO2 和CH3OH 合成DMC 的催化性能Table 6 Catalytic performance of transition metal oxide catalysts for the synthesis of DMC from CO2 and CH3OH
Wu 等[31]制备出磷酸改性V2O5催化剂,反应结果表明,随着H3PO4含量增加,CH3OH 转化率和DMC 选择性均增加。当CH3OH/CO2=500/250 mmol,催化剂用量为0.5 g,反应温度为140 ℃,P/V=0.20(摩尔比)时,DMC 选择性为92.12%,DMC 收率为4.50 mmol/g。磷酸改性是提高V2O5活性的有效方法,改性后催化剂中产生Brønsted酸,酸性增强,促进CO2活化。
2000 年,Tomishige 等[32]将ZrO2·xH2O 在115–800 ℃下焙烧3 h 制备ZrO2,研究表明,DMC 生成速率很大程度上取决于ZrO2结构,氢氧化锆在400 ℃焙烧时生成的亚稳态正方晶系ZrO2有利于DMC 生成;DMC 生成量随反应温度升高而增加,在170 ℃时达到最大值;CO2/CH3OH=250/192 mmol 时,DMC 生成量为0.33 mmol,且DMC生成速率几乎与CO2加入量成正比。此外,Tomishige等研究ZrO2吸附CO2和CH3OH 的FT-IR 谱图,提出CO2和CH3OH 合成DMC 的反应机理(图4),认为在酸性位点上CH3OH 活化(4)或中间产物CH3OC进一步反应生成DMC(5)为速率控制步骤。Jung等[33]采用原位红外光谱法研究了氧化锆上CO2和CH3OH 合成DMC 的机理,研究表明,CH3OH 在催化剂不饱和Zr4+Lewis 酸位点解离吸附要比CO2吸附慢得多。吸附时,CH3OH 的氧原子与催化剂表面的不饱和Zr4+阳离子结合,CH3OH 解离形成Zr-OCH3,并释放质子与表面羟基反应生成水,CO2插入Zr-OCH3上Zr-O 键中形成单齿碳酸甲酯基团(Zr-OC(O)OCH3),之后碳酸甲酯基团与CH3OH 反应形成DMC,并在氧化锆表面恢复羟基(Zr-OH)。
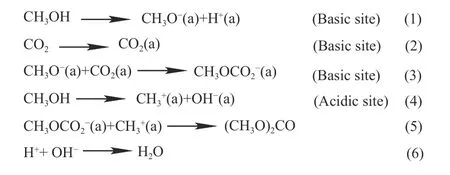
图4 ZrO2 上CO2 和CH3OH 合成DMC 反应机理[32]Figure 4 Reaction mechanism of synthesis of DMC from CO2 and CH3OH on ZrO2 [32](with permission from Elsevier)
2006 年,Yoshida 等[34]发现CeO2能催化CO2和CH3OH 合成DMC,并考察在不同煅烧温度下制备CeO2的催化性能。研究表明,在反应温度130 ℃和CO2初始压力1.2 × 106Pa 下,CH3OH 转化率为0.25%,DMC 选择性为100%。在600 ℃以上焙烧的催化剂,DMC 生成速率与其比表面积几乎成正比;随着焙烧温度升高,CeO2晶型由无定型态向萤石结构转变,且催化活性逐渐提高;结晶态CeO2比非晶态CeO2更利于DMC 合成。卢惠等[35]采用沉淀法制备CeO2催化剂,在最优条件下,DMC 收率为1.936 mmol/g;正交实验结果表明,硝酸铈溶液浓度对催化剂活性影响最为显著,其次是催化剂焙烧温度,KOH 溶液浓度影响最小。
Lee 等[36]考虑到催化剂酸性位点和碱性位点对该反应都有重要影响,通过溶胶凝胶法和浸渍法针对性制备出不同铈含量的CexZr1-xO2和MO/Ce0.6Zr0.4O2(MO=Ga2O3、La2O3、Ni2O3、Fe2O3、Y2O3、Co2O3、Al2O3)催化剂,在Ce0.6Zr0.4O2上负载MO 实现对催化剂的酸碱性调节。图5 为Ce0.6Zr0.4O2和MO/Ce1.6Zr0.4O2上CO2和CH3OH 合 成DMC 的催 化性能,负载MO 对CexZr1-xO2活性影响很大,Ga2O3/Ce0.6Zr0.4O2表现出最佳催化性能,酸碱位点和催化剂活性之间的协同机制首次被提出。由图6 可知,DMC 生成量随着催化剂酸性位点和碱性位点的增加而增加,且催化剂酸性和碱性显示出协同作用,两者在催化过程中不可或缺,催化剂酸碱位点数量和催化剂活性之间的相关机制如表7 所示。

图5 Ce0.6Zr0.4O2 和MO/Ce1.6Zr0.4O2 上CO2 和CH3OH 合成DMC 催化性能[36]Figure 5 Catalytic performance of Ce0.6Zr0.4O2 and MO/Ce1.6Zr0.4O2 for the synthesis of DMC from CO2 and CH3OH[36](with permission from Springer Nature)

图6 MO/Ce0.6Zr0.4O2 上酸碱位点和催化剂活性之间的协同机制[36]Figure 6 Synergy between the acid and basic sites for the catalytic activity of MO/Ce0.6Zr0.4O2 for the synthesis of DMC from CO2 and CH3OH[36](with permission from Springer Nature)
Tamboli 等[37]通过改进沉淀法制备更高比表面积(116 m2/g)的催化剂CexZr1-xO2用于DMC 合成,研究表明,Ce0.5Zr0.5O2表现出最佳催化活性,在CO2初始压力7.5 × 106Pa 和反应温度140 ℃下,CH3OH 转化率为4.93%,DMC选择性为100%。Kumar 等[38]通过模板法制备出一系列CexZr1-xO2催化剂。当x为0.5 时,催化剂Ce0.5Zr0.5O2上CH3OH转化率为0.77%,DMC 选择性为100%。
Li 等[39]通过溶胶凝胶法制备出不同铁含量固溶体催化剂FexZr1-xOy(x=0.3、0.5、0.7、0.9)。研究表明,随着催化剂中铁含量增加,催化剂比表面积也增大,催化剂Fe0.7Zr0.3Oy比表面积最高;在CO2压力1.2 × 107Pa 和反应温度110 ℃下测试催化剂活性,Fe0.7Zr0.3Oy上DMC 收率最高,这可能是高比表面积使更多活性位点暴露所导致。此外,Li 等还研究了催化活性与中等强度酸性位点和碱性位点数量的关系,如图7 所示。纯ZrO2和Fe2O3催化活性较低可能是由于表面酸性位点和碱性位点较少;Fe-Zr 固溶体催化剂表现出比纯ZrO2和Fe2O3更高的催化活性,可能是由于铁加入增加了催化剂中等强度酸性位点和中等强度碱性位点的数量,DMC 收率随它们增加而线性增加,这一结论与Lee 等[36]的结论相一致。
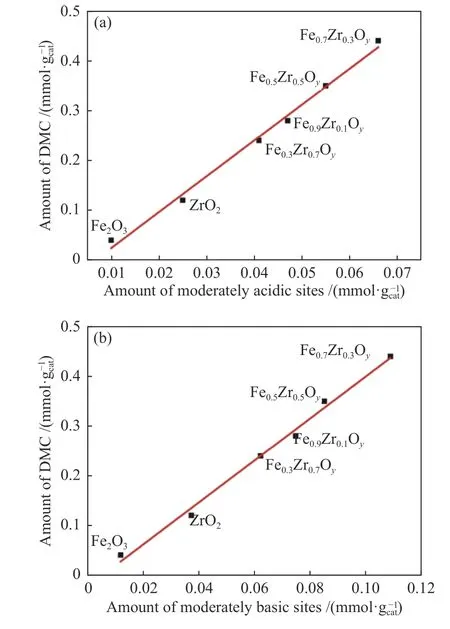
图7 (a)催化活性与催化剂中等强度酸性位点数量关系,(b)催化活性与催化剂中等强度碱性位点数量关系[39]Figure 7 Relationship between the catalytic activity and the number of acidic sites in the catalyst (a) as well as the relationship between the catalytic activity and the number of basic sites in the catalyst (b) in the synthesis of DMC from CO2 and CH3OH[39](with permission from Elsevier)
Kumar 等[40]通过共沉淀法制备CeO2催化剂,在反应温度120 ℃和CO2压力1.5 × 107Pa 下,反应4 h 后CH3OH 转化率为0.66%,DMC 选择性为100%。CeO2催化剂在连续五次循环使用后,DMC收率从2.046 mmol/g 下降到1.901 mmol/g,CH3OH转化率从1.81%下降到0.58%。Cui 等[41]考察煅烧气氛(空气、氧气、氢气、氩气)对纳米CeO2催化活性的影响。在不同煅烧气氛中,催化活性依次为:CeO2-氧气>CeO2-氩气>CeO2-空气>CeO2-氢气,CeO2-氧气上CH3OH 转化率为0.51%,DMC 选择性为100%。
2018 年,Fu 等[42]通过水热法制备Ti0.04Ce0.96O2纳米棒催化剂,在反应温度120 ℃下CH3OH 转化率和DMC 选择性分别为5.38%和83.1%。关于CO2和CH3OH 合成DMC 反应的速率控制步骤涉及两种不同机制[43],如表8 所示,Eley-Rideal(E-R)机制认为CO2与催化剂表面活化的MeOH*反应(S2)为速率控制步骤;Langmuir-Hinshelwood(LH)机制认为CO2和CH3OH 应在两个单独步骤中活化;关于L-H 机制的速率控制步骤存在争议,Tomishige 等[32]和Marin 等[44]分别将MeOH*形成步骤(S2)和CO2活化步骤(S1)视为速率控制步骤。为明确这一观点,Fu 等[42]以Ti0.04Ce0.96O2纳米棒为催化剂,采用初始速率法在固定床反应器中对该反应动力学进行研究。针对L-H 机制,将S3 假设为速率控制步骤,研究结果表明,物料浓度PCO2∙PCH3OH和初始速率呈现负线性相关,与理论结果不一致;将CO2吸附和活化作为速率控制步骤(S1),随反应物进料量变化,PCO2与初始速率之间呈正相关,初始速率方程为:k[CO2]-1[*]-1,这一结论与L-H 机制中以CO2活化为速率控制步骤相一致。此外,阿伦尼乌斯线性关系显示Ti0.04Ce0.96O2催化剂上表观活化能为46.3 kJ/mol,掺杂Ti 可以减少表观活化能,有利于DMC 合成。
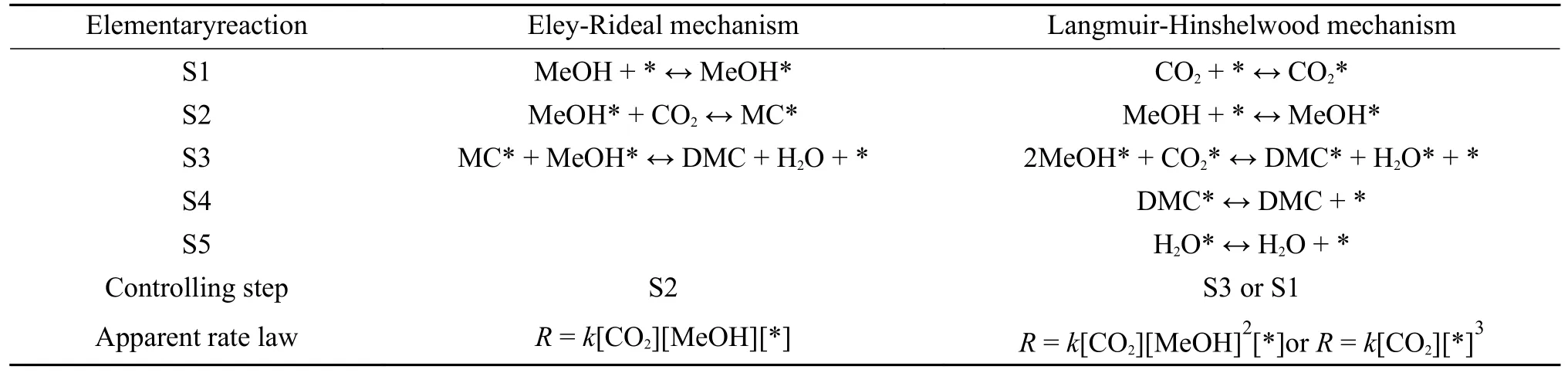
表8 基于Eley-Rideal 和Langmuir-Hinshelwood 的反应机制[33,37]Table 8 Reaction mechanisms for the synthesis of DMC from CO2 and CH3OH,based on the Eley-Rideal and Langmuir-Hinshelwood mechanisms[33,37]
近年来,Chen 等[45-48]将TixCe1-xO2、Ce1-xMgxO2、ZnxCe1-xO2和BixCe1-xOδ纳米复合材料涂敷在蜂窝状堇青石上制备成整体式催化剂,将其应用于CO2和CH3OH 合成DMC,在固定床反应器上测试其催化活性,如表6 所示。与前文提及的催化剂相比,CH3OH 转化率和DMC 收率明显提高,并且整体式催化剂具有稳定的机械强度和良好的回收利用活性,为CO2和CH3OH 合成DMC 的工业化提供了更大可能性。整体式催化剂在固定床中的应用如图8 所示。

图8 整体式催化剂结构及其在固定床反应器中的应用[48]Figure 8 Monolithic catalyst structure and its application in the synthesis of DMC from CO2 and CH3OH in a fixed-bed reactor[48](with permission from Royal Society of Chemistry)
迄今为止,金属氧化物催化剂主要是CeO2、ZrO2、V2O5和其他过渡金属氧化物,其CH3OH 转化率很难超过5%,但DMC 选择性很容易达到100%。在固定床反应器中,铈基复合氧化物催化剂上CH3OH 转化率达到20%以上,DMC 选择性在80%左右。
2.4 Cu-Ni 合金催化剂
Cu-Ni 合金催化剂为CO2和CH3O 合成DMC研究热点,Cu-Ni 双金属作为活性组分,需要借助一些具有较大比表面积和足够机械强度的载体来充分发挥其催化性能。
近年来,用于CO2和CH3OH 合成 DMC 的Cu-Ni双金属相催化剂和载体主要包括Cu-Ni/ VSO(V2O5-SiO2)、Cu-Ni/MWCNT(多壁碳纳米管)、Cu-Ni/HNTs(K 改性埃洛石纳米管)等,其催化活性如表9 所示。

表9 Cu-Ni 合金催化剂上CO2 和CH3OH 合成DMC 催化性能Table 9 Catalytic performance of the Cu-Ni alloy for the synthesis of DMC from CO2 and CH3OH
2006 年,Wu 等[49]制备出Cu-Ni/VSO,在140 ℃和9.0 × 105Pa 下催化CO2和CH3OH 合成DMC,CH3OH 转化率为2.4%,DMC 选择性为87.2%。FT-IR 结果显示,当V2O5负载在SiO2上时,形成新的V-O-Si 键;DRIFT 光谱表明,CO2和CH3OH 被吸附到催化剂表面活化,这对于催化反应至关重要。
2009 年,Bian 等[50]制备出双金属催化剂Cu-Ni/MWCNT(多壁碳纳米管),在图9 所示的固定床反应器中测试催化活性。图10 为催化剂稳定性测试,结果表明,在10 h 内,CH3OH 转化率从4.44%缓慢下降到4.32%,DMC 选择性从91.0%下降到90.8%;在0.5–2.5 h,催化活性增加,可能是由于在反应初始阶段形成了活性物质;在2.5–7 h,DMC 收率逐渐降低;反应7 h 后,DMC 收率趋于平衡。
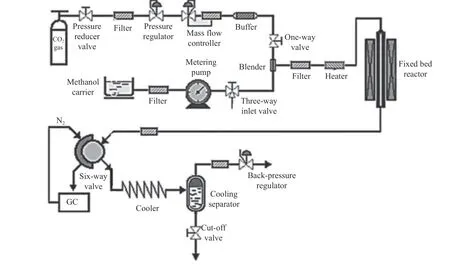
图9 Cu-Ni/MWCNT 上连续合成DMC 装置示意图[50]Figure 9 Schematic diagram of a device for the continuous synthesis of DMC from CO2 and methanol on the Cu-Ni/MWCNT catalyst[50](with permission from Elsevier)

图10 Cu-Ni/MWCNT 催化剂稳定性研究[50]Figure 10 Stability test of the Cu-Ni/MWCNT catalysts for the synthesis of DMC from CO2 and CH3OH[50](with permission from Elsevier)
埃洛石纳米管(HNT)具有大量酸中心[51],已被用作多种反应的酸催化剂,并且HNT 的亲水性[52]能使水和CH3OH 的混合物分离。Zhou 等[53]以HNT为载体制备催化剂Cu-Ni/HNTs,并对其K 改性制备Cu-Ni/KHNTs。研究发现,当KHNTs 上金属负载量为21%时,CH3OH 转化率为7.85%;金属负载量为3%和21%的催化剂具有更多酸性位点或碱性位点,表现出更高催化活性;金属负载量为9%和15%的催化剂表面上存在相近摩尔量的酸性位点和碱性位点,催化活性较差,这可能是由于反应物活化竞争所导致,对DMC 形成不利。催化剂酸碱性质可以通过改变催化剂组成来调节,酸碱位点平衡是CO2和CH3OH 生成DMC的原因。
Bian 等[54]考虑到氧化石墨烯(GO)优异的电子传输性能、高电导率和亲水性,将其作为载体制备Cu-Ni/GO 催化剂。实验表明,随着金属负载量增加,DMC 收率普遍提高,当金属含量为20%,在最优条件下,DMC 收率为9.0%。Cu-Ni/GO 催化剂上CO2和CH3OH 合成DMC 反应机理如图11所示,M 表示Cu、Ni 或Cu-Ni 合金,反应步骤可概括为:(1)CH3OH 在催化剂表面M 上被活化形成CH3O-;(2)CO2在催化剂表面被活化形成-C=O;(3)CH3O–和-C=O 结合形成碳酸甲酯中间体,该中间体再与另一CH3O–结合生成DMC。石墨的低碳电负性使电子易于释放,这是CO2活化关键,电子转移在反应物活化和DMC 形成中发挥重要作用。
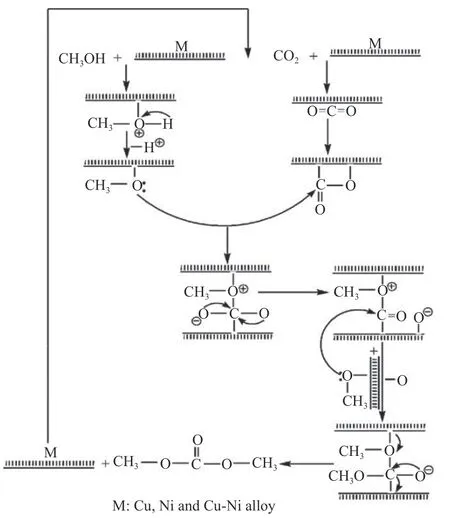
图11 Cu-Ni/GO 上CO2 和CH3OH 合成DMC 反应机理[54]Figure 11 Reaction mechanism of synthesis of DMC from CO2 and CH3OH on Cu-Ni/GO[54](with permission from Elsevier)
Chen 等[55]制备出分子筛负载Cu-Ni 合金催化剂,在固定床反应器中测试催化活性,DMC 收率为5.0%。Bian 等[56]制备出20%Cu-Ni/活性炭(AC)和钒掺杂Cu-Ni/VAC 催化剂。研究发现,钒掺杂能有效提高催化剂活性和选择性。在最佳条件下,Cu-Ni/VAC 催化剂上CH3OH 的转化率为7.76%,DMC 选择性为89.9%;而Cu-Ni/AC 催化剂上CH3OH转化率仅为6.44%,在所有反应温度下,钒掺杂Cu-Ni/AC 催化剂活性均明显提高。
Zhang 等[57]采用硫化法和传统溶液法制备出Cu-Ni/VSiO 催化剂。结果表明,硫化法制备的Cu-Ni/VSiO 上CH3OH 转化率为4.2%,DMC 选择性为93.1%,比溶液法制备的催化剂CH3OH 转化率高2.7 倍。
Cu-Ni 合金催化剂相比于其他类型催化剂,其性能有所提升,CH3OH 转化率在4%–10%,但很难超过10%,DMC 选择性在90%左右。活性组分容易氧化失活,催化剂长期储存是关键问题。
2.5 MOFs 催化剂
MOFs 是一种多孔结晶材料,由金属离子或金属簇与有机配体配位而成的网状结构晶体,它的高比表面积、大孔隙率以及可调节的金属中心使其成为有吸引力的催化剂。
2017 年,Verma 等[58]首次使用MOFs 催化CO2和CH3OH 合成DMC,设计制备出多种MOFs催化剂(简要制备流程如图12 所示)。在反应温度100 ℃和CO2压力2.07 × 105Pa 下,催化剂Ti-ZTBF 上DMC 收率达到16%。Ti-ZTBF 的多孔形态和高比表面积使反应能够在较低温度和CO2压力下进行。

图12 不同配体和钛基沸石咪唑酸盐框架的合成[58]Figure 12 Synthesis of different ligands and Ti-based zeolitic imidazolate frameworks[58]
Poungsombate 等[59]通过水热法制备出沸石咪唑框架-8 载体(ZIF-8),采用浸渍法制备Cu-Ni/ZIF-8,研究金属负载量、反应温度、催化剂用量和反应时间等参数对反应的影响,结果表明,催化剂5%Cu-Ni/ZIF-8 用 量0.7 g、CO2压 力2.0 × 106Pa、反应温度110 ℃,反应12 h 后CH3OH 转化率为12.79%,DMC 收率为6.39%。
2018 年,Xuan 等[60]制备出一系列MOFs 催化剂UiO-66-X(X为三氟乙酸TFA 与对苯二甲酸BDC的摩尔比,X=0、6、12、18、24),结果表明,UiO-66-24 具有最高比表面积(1479 m2/g)和最多催化活性位点(8.3 mmol/g),该催化剂上DMC 收率为0.084%。此外,Xuan 等[61]制备出催化剂MOF-808-X(X为ZrOCl2∙8H2O 与1,3,5-苯三甲酸BTC 的物质的量比)。结果表明,MOF-808-4 具有最高比表面积(1373 m2/g)和最多催化活性位点(9.8 mmol/g),该催化剂上DMC 收率为0.12%。尽管UiO-66-24 具有更高比表面积,但是其上DMC 收率比MOF-808-4 上的低,这可能是由于MOF-808-4 的微孔尺寸比较大,使得反应物更容易接近微孔中的活性位点。
此外,Xuan 等[61]提出MOF-808-4 催化剂上的反应机理,如图13 所示,可概括为:(1)CH3OH 首先与MOF-808-4 中Zr6节点处Lewis 酸位点(Zr4+)结合形成Zr–OCH3,并释放H 质子,H 质子随后与末端羟基Zr–OH 迅速反应生成H2O;同时,CH3OH 还可以与Zr6节点处碱性位点(Zr–O–Zr 上的不饱和O2–或者Zr–O–)结合形成桥接甲氧基Zr–(OCH3)–Zr,CH3OH 中解离的羟基可以吸附在酸性位点Zr4+上形成末端羟基Zr–OH;(2)Zr6节点处碱性位点上吸附的CO2插入到Zr–OCH3中形成中间产物Zr–OCOOCH3;(3)Zr–(OCH3)–Zr 释放出的CH3+与Zr–OCOOCH3结合生成DMC。
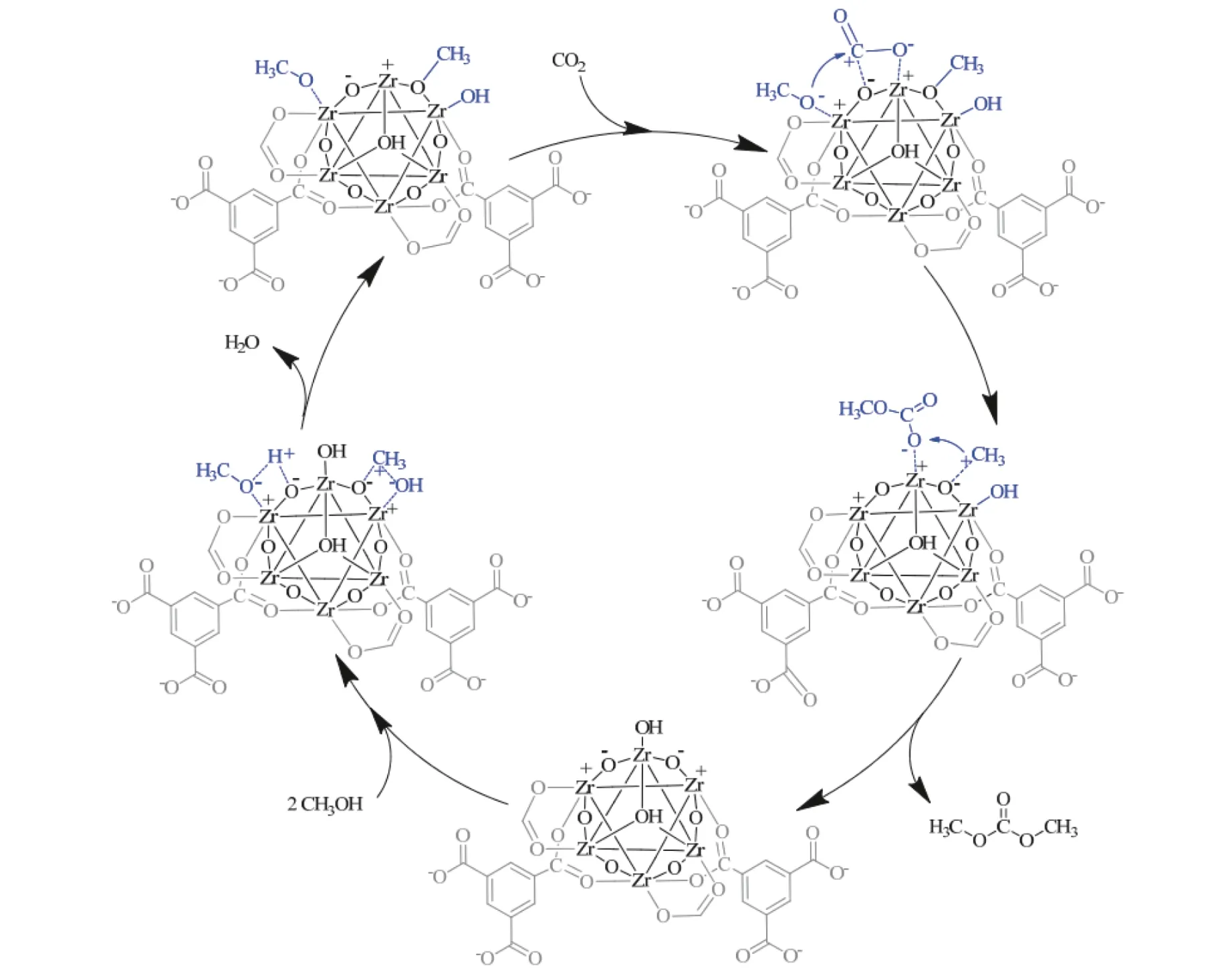
图13 MOF-808-4 上CO2 和CH3OH 合成DMC 反应机理[61]Figure 13 Reaction mechanism of synthesis of DMC from CO2 and CH3OH on MOF-808-4[61](with permission from Elsevier)
根据Jung 等[33,62,63]报道的ZrO2催化CO2和CO3OH 合成DMC 反应机理,认为在DMC 合成过程中CHC的生成为速率控制步骤,而在MOF-808-4 催化剂上Zr–(OCH3)–Zr 上C–O 键可以快速断裂形成 C,这一结果可能归因于MOF-808-4 上Zr6节点处酸性位点和碱性位点处于相邻位置,Tomishige 等[32]和Jung 等[33]已经揭示ZrO2中相邻酸性位点(暴露的Zr4+和羟基)和碱性位点(Zr–O–Zr中的不饱和O2–或Zr–O–)有利于Zr–(OCH3)–Zr中C–O 键断裂形成 C,以及 C与碳酸甲酯物种结合形成DMC。
2020 年,Kumar 等[64]利用机械法和水热法制备出Cu2S 键合磺化有机聚合物催化剂Cu-SOPm和Cu-SOPs。在反应温度110 ℃和CO2初始压力3.0 × 106Pa 下,在10 mL 反应釜中反应2 h,Cu-SOPm和Cu-SOPs 上DMC 收率分别为18.3%和11.7%。之后在固定床反应器中测试Cu-SOPm 的催化活性,在反应温度130 ℃时,DMC 收率为23.2%。将反应混合物连续反应两次,在首次连续运行中,DMC 收率达到42.8%,无任何副产物,第二次运行后DMC 的总收率为53.1%,但是选择性下降,可能原因是水饱和导致DMC 的分解。此外,Kumar等[64]提出Cu-SOPm 催化剂上的反应机理,如图14所示,涉及三个基本步骤:(1)CO2在碱性位点(–SO3H和–SO2H)上活化并通过Cu+物种(–Cu2S)稳定存在;(2)CH3OH 在酸性位点和碱性位点活化分别形成 C和CH3O–;(3)C和CH3O–与活化的CO2分子反应生成DMC。催化剂表面Brønsted 酸性位点–SO3H 和–SO2H 吸附反应中形成的水,并且发现水比CH3OH 优先吸附在催化剂表面;Cu-SOPm水蒸气吸附量高于Cu-SOPs,因为其表面存在更多–SO3H 基团,说明磺化能改善催化剂亲水性,有利于水分子在催化剂上优先吸附。
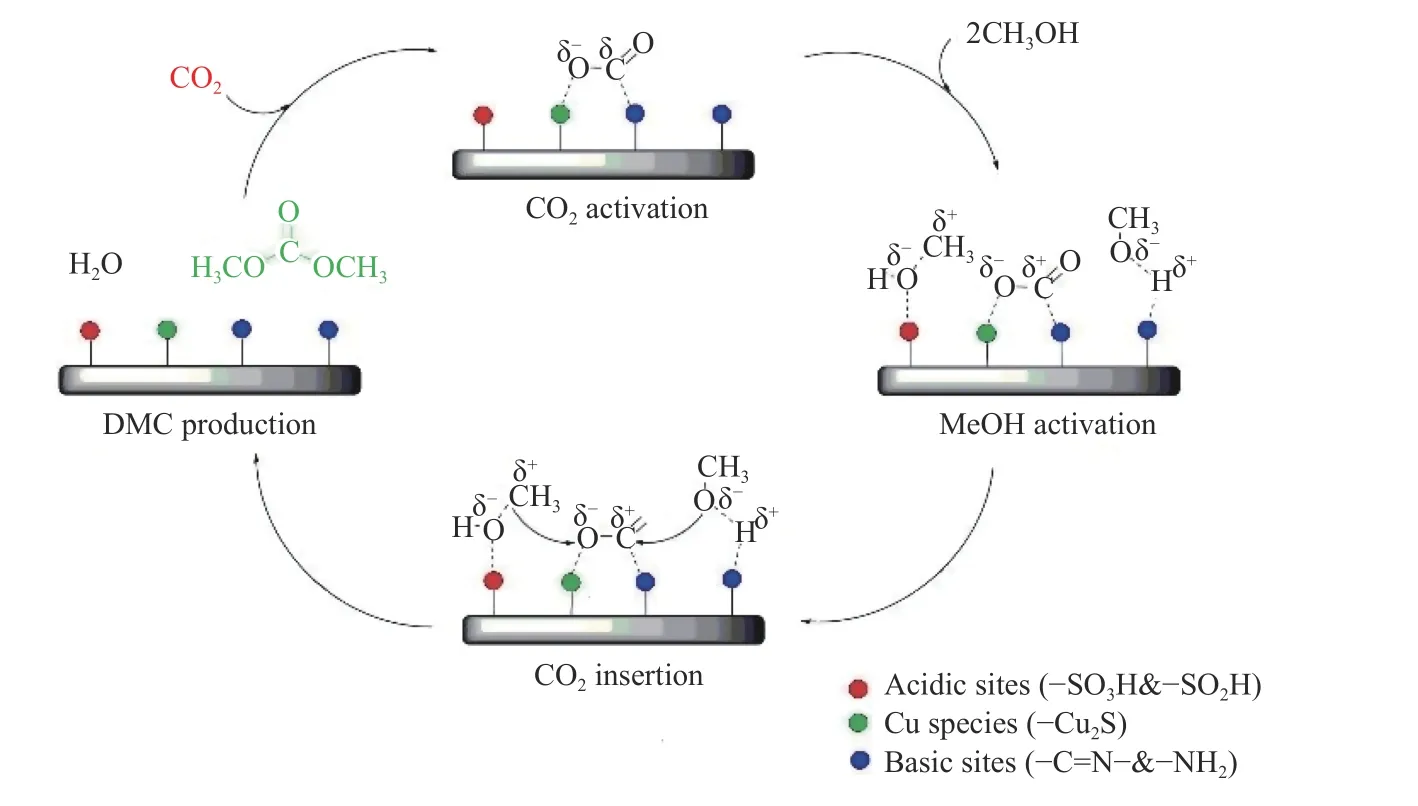
图14 Cu-SOPm 上CO2 和CH3OH 合成DMC 反应机理[64]Figure 14 Reaction mechanism of synthesis of DMC from CO2 and CH3OH on Cu-SOPm[64](with permission from Royal Soc of Chem)
Jiang 等[65]制备出催化剂Ce-MOF-T-H/N/A,T为煅烧温度,H/N/A 分别代表氢气、氮气和空气三种煅烧气氛。在反应温度140 ℃,CO2初始压力5.0 × 106Pa 下测试催化剂活性,结果表明,Ce-MOF-500 ℃-H 的催化活性最高,DMC 收率为6.332 mmol/g,这主要是由于在H2气氛中煅烧得到的催化剂比表面积大(71.65 m2/g)和氧空位含量高。
2.6 杂多酸催化剂
杂多酸是由杂原子(如P、Si、Fe、Co 等)和多原子(如Mo、W、V、Nb、Ta 等)按一定结构通过氧原子配位桥联组成的一类含氧多酸,具有特殊空间结构和较高催化活性,是一种多功能新型催化剂,在催化研究领域中受到研究者广泛重视。表10 为近年来杂多酸上CO2和CH3OH 合成DMC催化活性。
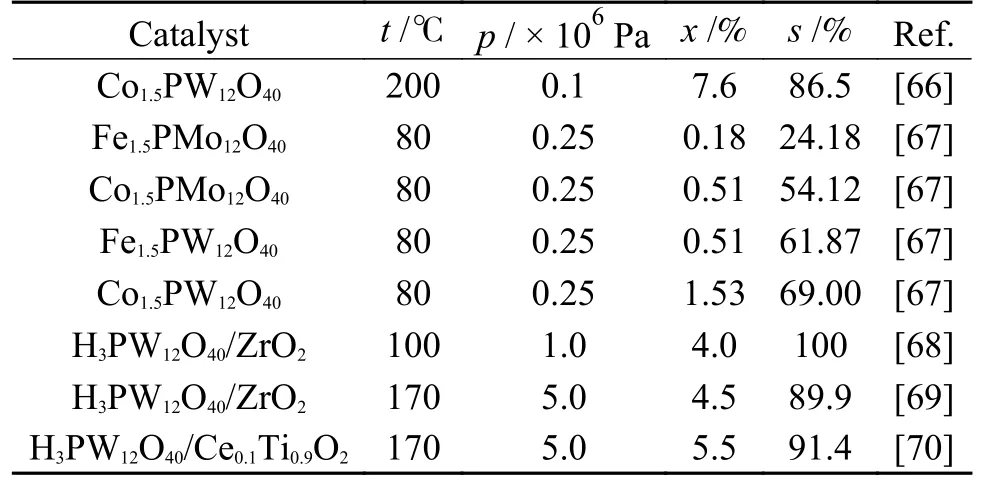
表10 杂多酸催化剂上CO2 和CH3OH 合成DMC 催化性能Table 10 Performance of the heteropolyacid catalysts for the synthesis of DMC from CO2 and CH3OH
Aouisi 等[66,67]对Keggin 型杂多酸催化剂进行研究,并将其应用于CO2和CH3OH 合成DMC 反应中。2010 年,Aouisi 等[66]制备出Keggin 型杂多阴离子催化剂Co1.5PW12O40,研究反应温度、反应压力和CH3OH 质量空速(MWHSV)对CH3OH 转化率和DMC 选择性的影响。结果表明,在200 ℃、常压、3.2 h-1下,CH3OH 转化率为7.6%,DMC 选择性为86.5%。CH3OH 转化率和DMC 选择性随反应温度升高而降低,这是因为在较高反应温度下CO2在催化剂表面吸附量降低。Aouisi 等[66]还研究了具有不同杂原子(Co 和Fe)和多原子(W 和Mo)的Keggin 型杂多酸催化剂,与其他催化剂相比,Co1.5PW12O40的催化活性最高,可能是因为Co原子和W 原子的协同作用。
此外,Jiang 等[68]通过溶胶凝胶法制备H3PW12O40/ZrO2催化剂,在反应温度100 ℃和CO2压力1.0 × 106Pa 下,CH3OH 转化率为4.0%,DMC选择性为100%。CO2和CH3OH 在H3PW12O40/ZrO2上合成DMC 反应机理,如图15 所示,(1)当CH3OH吸附在H3PW12O40/ZrO2表面时,CH3OH 中羟基与催化剂碱性位点结合,t-OH 和b-OH 转化为末端甲氧基t-OCH3和桥接甲氧基b-OCH3;(2)在催化剂碱性位点上,t-OCH3与CO2反应生成碳酸甲酯阴离子,b-OCH3不参与反应;(3)CH3OH 在Brønsted酸性位点活化为 C和OHδ-,碳酸甲酯阴离子与甲基阳离子反应形成DMC。此外,Jiang等认为H3PW12O40/ZrO2上合成DMC 的高选择性可能是由于在反应过程中t-OCH3快速转化为碳酸甲酯物种;基本反应步骤(3)是该反应的速率控制步骤;H3PW12O40/ZrO2表面Brønsted 酸位点在CH3OH 活化中比Lewis 酸位点发挥着更重要的作用。
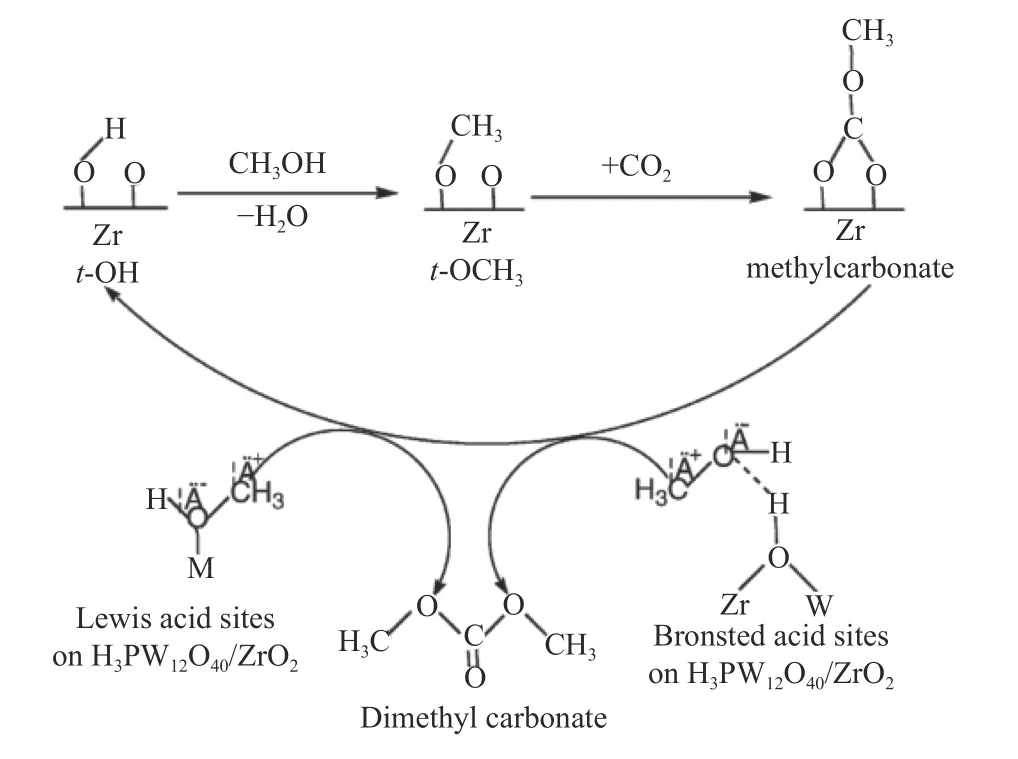
图15 H3PW12O40/ZrO2 上CO2 和CH3OH 合成DMC反应机理,M=W 或Zr[68]Figure 15 Reaction mechanism of synthesis of DMC from CO2 and CH3OH on the H3PW12O40/ZrO2 catalysts (M=W or Zr)[68](with permission from Elsevier)
Chiang 等[69]以H3PW12O40/ZrO2为催化剂,在反应温度170 ℃、CO2压力5.0 × 106Pa 和CO2/N2体积流率比1∶7 下,CH3OH 转化率和DMC 选择性分别为4.5%和89.9%。此外,伪一级线性回归模型和阿伦尼乌斯线性关系图结果显示,反应最佳速率常数为4.24 × 10-3min-1,表观活化能为15.54 kJ/mol,这些数据为CO2和CH3OH 合成DMC 理论计算提供了重要补充。
2017 年,Chiang 等[70]通过溶胶凝胶法制备H3PW12O40/Ce0.1Ti0.9O2催化剂,在反应温度170 ℃、CO2压力 5.0 × 106Pa 和CO2/N2=1∶7 的体积流率比下,CH3OH 转化率和DMC 选择性分别为5.5%和91.4%。H3PW12O40/Ce0.1Ti0.9O2上CO2和CH3OH合成DMC 反应机理如图16 所示,H3PW12O40/Ce0.1Ti0.9O2催化剂表面氧空位作为反应Lewis 酸位点,CO2分子中氧原子插入氧空位,另一氧空位被CH3OH 中氢原子填充;同时,又一CH3OH 分子吸附在相邻的氧空位上,两分子CH3OH 和一分子CO2结合形成不稳定中间体;最后,该中间体分解为DMC 和H2O 分子,它们从氧空位上脱附,完成催化循环。该反应机理从氧空位和晶体缺陷角度解释了杂多酸催化本质。
目前,杂多酸催化剂主要包括Co1.5PW12O40、H3PW12O40/ZrO2、H3PW12O40/Ce0.1Ti0.9O2等,这些催化剂上CH3OH 转化率很难超过10%,DMC 选择性容易超过90%。
3 脱水对CO2 和CH3OH 直接合成DMC 的影响
根据过去几十年文献报道,脱水对CO2和CH3OH合成DMC 反应有非常重要的影响,主要是因为原位水脱除有助于化学平衡右移,从而增加CH3OH转化率和DMC 收率,且在反应过程中由于水存在会使催化剂中毒从而迅速降低活性。本工作针对世界各国效果较好的物理脱水剂、化学脱水剂和改进的脱水反应器等对该反应的影响进行归纳总结。
3.1 2-氰基吡啶脱水剂
在2014 年,Bansode 等[71]在固定床反应器中(图17)以2-氰基吡啶(2-CP)为脱水剂,在CeO2催化剂上连续合成DMC。向反应器中不断通入CO2和一定化学计量比的CH3OH 和2-CP 混合物,在较宽压力范围下操作。研究表明,在最优反应条件下,液相产物中CH3OH 转化率在95%以上,DMC 选择性接近100%。如表11 所示,为近年来以2-CP 为脱水剂的不同催化-脱水体系中CO2和CH3OH 合成DMC 性能。

图17 DMC 连续合成工艺流程示意图[71]Figure 17 Schematic diagram of continuous DMC synthesis process[71](with permission from ACS Publications)
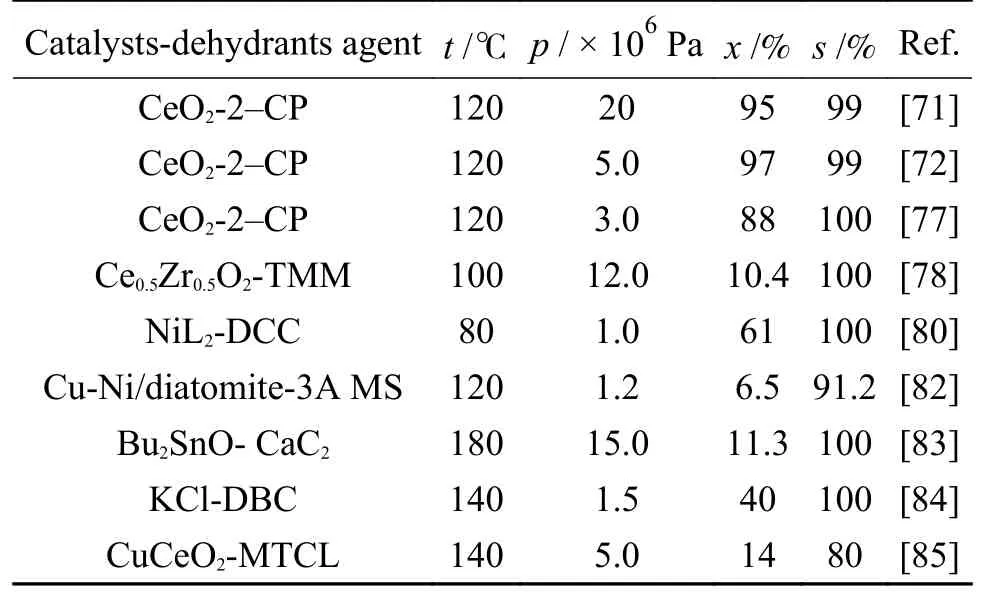
表11 不同催化-脱水体系中CO2 和CH3OH合成DMC 性能Table 11 Performance of various catalytic-dehydration systems for the synthesis of DMC from CO2 and CH3OH
Honda 等[72]研究2-CP 参与下CO2和CH3OH合成DMC 体系中的主要反应路线,如图18 所示,2-CP 水合作用产生2-吡啶酰胺(2-PA),该反应也由CeO2催化[73],2-PA 也可以在Na2O/SiO2催化剂上脱水还原为2-CP[74,75];副产物吡啶甲酸甲酯、氨基甲酸甲酯和吡啶-2-甲酰亚胺甲酯分别通过式(3)、(4)、(5)产生。此外,Honda 等[72]提出CeO2-2-CP 催化体系中CO2和CH3OH 合成DMC 反应机理,如图19 所示,(A)为CH3OH 活化过程,(ⅰ)CH3OH 吸附在CeO2上形成CH3O-Ce;(ⅱ)CO2插入CH3O-Ce 中生成碳酸甲酯;(ⅲ)CH3OH 吸附在CeO2催化剂表面形成CH3O-Ce;(ⅳ)CH3O-Ce以亲核加成的方式与碳酸甲酯结合生成DMC(通过18O 标记甲醇参与反应证实),CeO2再生;(B)为2-CP 水合过程,(ⅵ)2-CP 中吡啶环N 原子与Ce-OH结合形成2-PA;(ⅶ)、(ⅷ)由于2-PA 酸强度较小,酰胺基中H 原子和吡啶环N 原子之间形成分子内氢键[76],使2-PA 在CeO2上吸附较弱,2-PA 可以从CeO2表面快速脱附;(ⅴ)反应体系中的水可以很容易地在CeO2上解离为Ce-OH 和H 质子。反应生成DMC 速率控制步骤是甲氧基亲核加成到CeO2表面碳酸甲酯中间体上;2-CP 与Ce-OH 结合形成2-PA 是2-CP 水合过程的速率控制步骤[73]。另外,动力学研究表明,2-CP 存在不会改变CO2和CH3OH 合成DMC 反应机理。
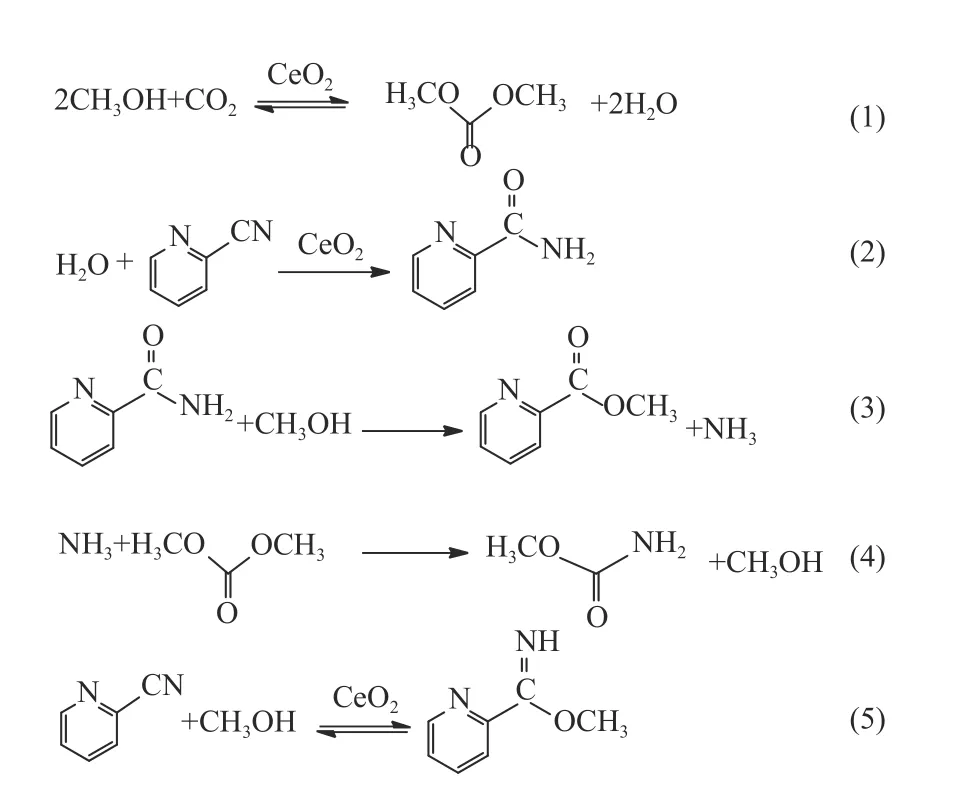
图18 CeO2-2–CP 催化CO2 和CH3OH 合成DMC 反应体系研究[73]Figure 18 Reaction system of synthesis of DMC from CO2 and CH3OH catalyzed by CeO2-2–CP[73](with permission from Elsevier)

图19 CeO2-2-CP 催化体系中CO2 和CH3OH 合成DMC 反应机理[72]Figure 19 Reaction mechanism of synthesis of DMC from CO2 and CH3OH in the CeO2-2–CP catalytic system[72](with permission from Elsevier)
2018 年,Stoian 等[77]认为,2-CP 水合产生2-PA 导致CeO2催化剂表面中毒,如图20 所示,(a) CH3OH分子以t-OCH3和b-OCH3的形式吸附在CeO2上;(b) 2-CP 到达催化剂表面时,与CH3OH 竞争吸附,在氧空位上吸附水解形成中间体2-PA(深红色),占据CH3OH 活性位点。为增加CeO2催化剂稳定性,通过在CeO2催化剂中添加1%稀土金属(La、Gd 和Pr)对CeO2进行表面改性,结果表明,1%Pr 和1%Gd 对CeO2促进作用更加明显。在最佳条件下运行150 h,与单独使用CeO2相比,使用1%Pr-CeO2和1%Gd-CeO2催化剂DMC 收率提高35%。ATR-IR 光谱表明,稀土金属能够增强甲氧基物质吸附强度从而占据合成DMC 活性位点,阻止2-PA 吸附,在一定程度上避免催化剂失活。
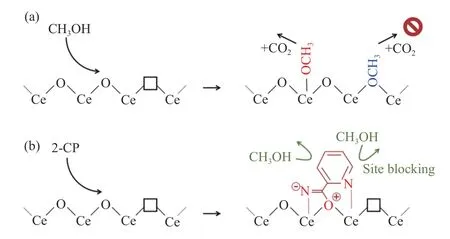
图20 2-CP 对CeO2 催化剂表面活性位点的影响[77]Figure 20 Effect of 2-CP on the surface active sites of the CeO2 catalyst [77](with permission from ACS Publications)
3.2 其他脱水剂
2011 年,Zhang 等[78]考 察Ce0.5Zr0.5O2-TMM(1,1,1–三甲氧基甲烷)催化体系对CO2和CH3OH 合成DMC 影响。研究表明,在反应温度100 ℃和CO2压力1.2 × 107Pa 下,与没有脱水剂参与相比,甲醇转化率从1.8%提升到10.4%,DMC 选择性为100%。TMM 参与下,反应机理如图21 所示,TMM不仅可以和体系中H2O 结合生成主反应物CH3OH,还可作为原料,在高压条件下与超临界CO2结合生成DMC,但TMM 价格昂贵且难以再生利用。

图21 TMM 促进CO2 和CH3OH 合成DMC 反应机理[78]Figure 21 Reaction mechanism of synthesis of DMC from CO2 and CH3OH promoted by TMM[78](with permission from ACS Publications)
2005 年,Aresta 等[79]以CuCl 为催化剂,以二环己基碳二亚胺(DCC)为脱水剂,在反应温度65 ℃和CO2压力5.0 × 106Pa 下反应6 h,DMC 收率为30.2%,反应时间延长至24 h,DMC 收率为83.3%。催化反应机理如图22 所示,(Ⅰ) CH3OH 首先进攻DCC 中双键碳原子,形成O-甲基异脲中间体;(Ⅱ)CH3OH 分子与O-烷基异脲中间体以氢键方式连接形成中间体6aN-im;(Ⅲ) CO2与中间体6aN-imN结合形成中间体13a,该过程分两步进行:第一步,在CO2促进下,6aN-imN 中质子从CH3OH 转移到异脲中亚氨基氮上,其与甲氧基阴离子相互作用形成“离子对”中间体12a;第二步,异脲中带正电荷的甲基与碳酸甲酯阴离子-OC(O)OCH3结合形成中间体13a;(Ⅳ)中间体13a 形成DMC 和二环己基脲。研究表明,基本步骤(Ⅰ)可由CuCl、ZnCl2、CuO等催化,反应在60 ℃具有良好的选择性和收率;基本步骤(Ⅱ)、(Ⅲ)异脲与CH3OH 和CO2反应不需要任何催化剂,反应在温度60 ℃和CO2压力1.6 × 105Pa 下进行具有较高选择性和收率。
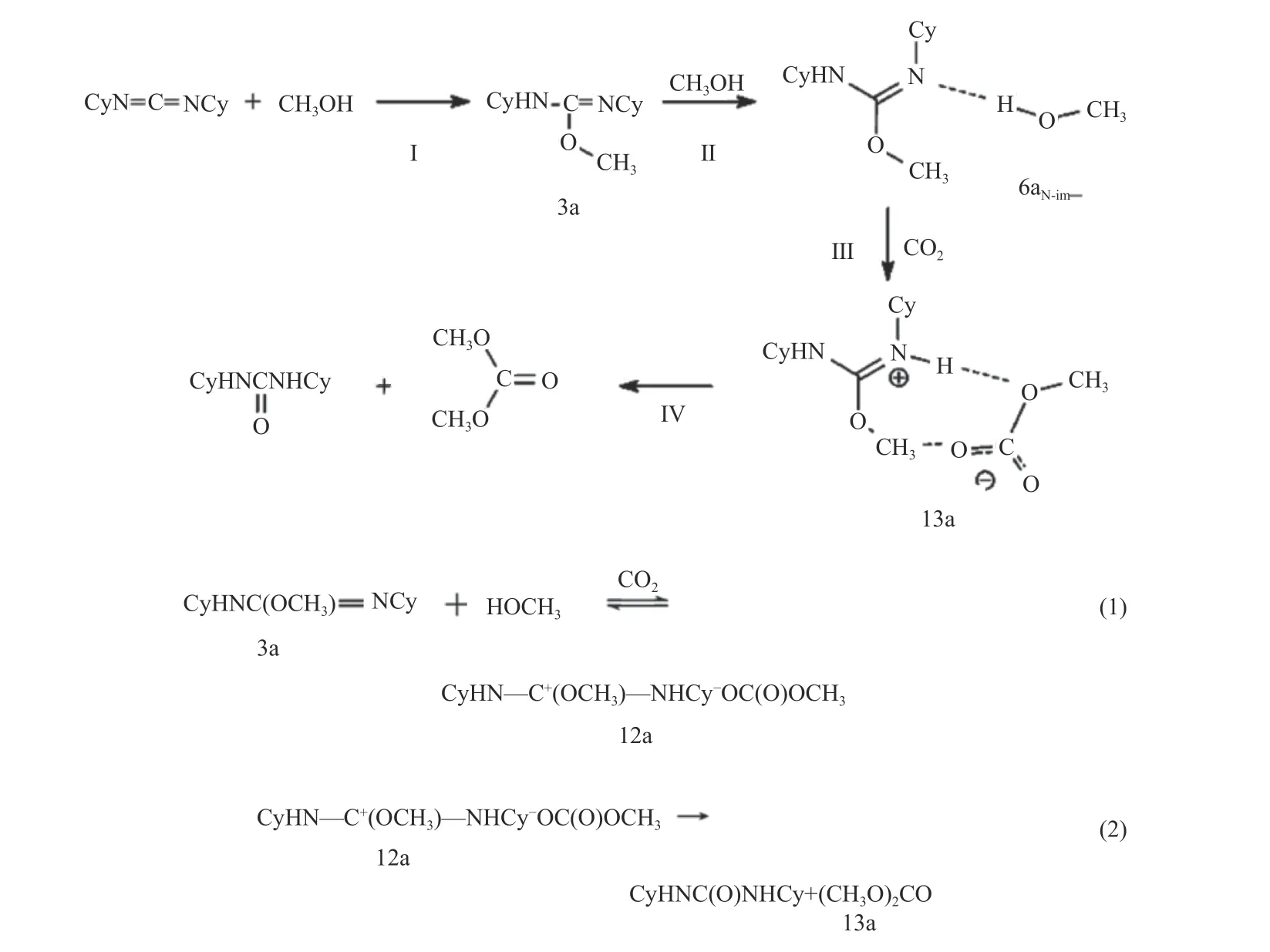
图22 DCC 促进CO2 和CH3OH 合成DMC 反应机理[79]Figure 22 Reaction mechanism of synthesis of DMC from CO2 and CH3OH promoted by DCC[79](with permission from ACS Publications)
2014 年,Shi 等[80]制备出催化剂双[2-(1H-苯并咪唑-2-基)苯甲酸]镍(II)(NiL2),以DCC 为脱水剂,在CO2压力1.0 × 106Pa 和反应温度80 ℃下,CH3OH 转化率为61%,DMC 选择性为100%。催化剂可以通过简单过滤、洗涤和干燥完成回收,重复使用五次后,DMC 收率从61%下降至58%。DCC与NiL2协同催化反应机理如图23 所示,CH3OH分子首先与NiL2发生吸附作用,CO2分子插入Ni–O 键之间形成碳酸甲酯中间体;同时,另一分子CH3OH 与DCC 中的碳氮双键加成,形成O-甲基异脲中间体,再与碳酸甲酯中间体反应生成DMC。NiL2在单独使用时不显示催化活性,必须配合DCC 完成。
2016 年,Wang 等[81]考察2,2-二甲氧基丙烷(DMP)脱水剂对不同形貌CeO2催化CO2和CH3OH合成DMC 的影响。研究表明,在没有DMP 参与时,在反应温度140 ℃和CO2初始压力5.0 × 106Pa下测试纺锤体CeO2、八面体CeO2、六面体CeO2的催化活性,DMC 收率分别为8.4、3.0、1.5 mmol/g;在相同条件下,在体系中加入DMP3.6 mL,三种催化剂DMC 收率分别为28.2、8.3、2.8 mmol/g。脱水剂DMP 的使用能使DMC 收率提升两倍左右。
2018 年,Han 等[82]制备出K 改性Cu-Ni 硅藻土纳米催化剂,在固定床反应器中考察3A 分子筛对CO2和CH3OH 合成DMC 反应的脱水效率,与未加分子筛相比,DMC 收率提高13%。与2-CP、DCC、DMP 等化学脱水剂相比,分子筛易于回收,不会产生副产物。
Zhang 等[83]以Bu2SnO 为催化剂用于CO2和CH3OH 合成DMC,研究电石(CaC2)与体系中水反应以改变反应平衡的能力。在Bu2SnO-CaC2体系中,在反应温度180 ℃和CO2压力1.5 × 107Pa 条件下,CH3OH 转化率为11.3%,DMC 选择性为100%。在系统中加入CaC2后,CH3OH 转化率从0.9%增加到11.3%。CaC2驱动CO2和CH3OH 合成DMC反应途径如图24 所示,在反应过程中生成的H2O被CaC2消耗产生C2H2气体,导致反应平衡移动。然而,CaC2不能重复使用。
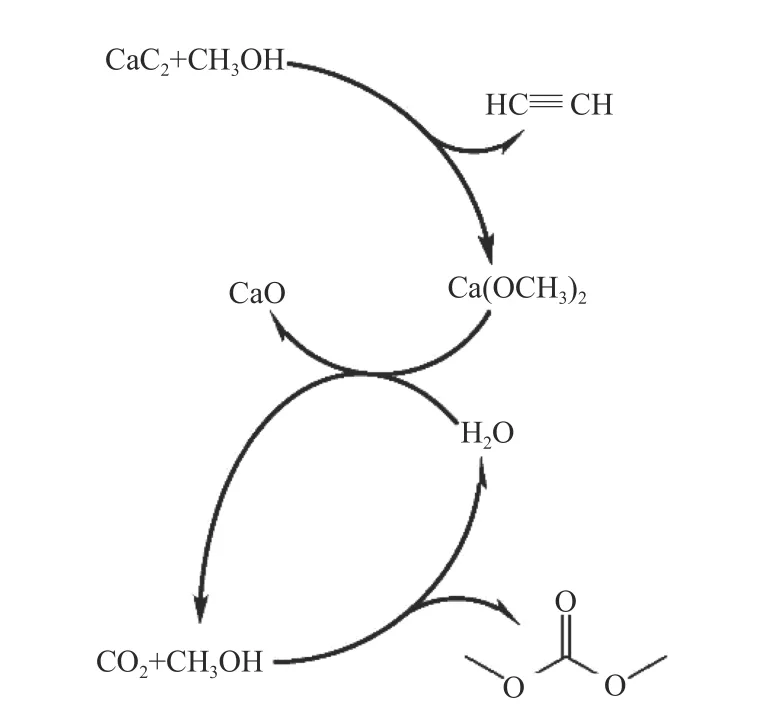
图24 CaC2 促进CO2 和CH3OH 合成DMC 反应机理[83]Figure 24 Reaction mechanism of synthesis of DMC from CO2 and CH3OH promoted by CaC2[83](with permission from Royal Soc of Chem)
Li 等[84]发现,冠醚对碱金属卤化物催化剂表现出共催化作用。以碱金属卤化物为催化剂,研究二苯并-18-冠醚-6(DBC)、18-冠醚-6、15-冠醚-5 和12-冠醚-4 对反应的促进作用。在CH3OH/环氧丙烷(PO)=7∶3(物质的量比),CO2压力1.5 ×106Pa 和反应温度140 ℃条件下,KCl-DBC 催化体系中CH3OH 转化率为40%,DMC 选择性为100%,与纯KCl 相比,CH3OH 转化率上升五倍以上。DBC可以通过形成有机金属络合物防止副反应发生,但是PO 的使用对CO2和CH3OH 合成DMC 成本增加,限制工业化应用。
2019 年,Marciniak 等[85]考察脱水剂三氯乙酸甲酯(MTCL)和2-CP 对CO2和CH3OH 合成DMC的影响。结果表明,在无脱水剂参与下,在反应温度140 ℃和CO2初始压力5.0 × 106Pa 条件下,0.02%CuCeO2催化剂上CH3OH 转化率为1.35%,DMC 选择性为100%。在上述相同条件下,加入等量MTCL 和2-CP,前者CH3OH 转化率和DMC选择性分别为14.0%和80%,后者为5.0%和100%。MTCL 脱水机理如图25 所示,MTCL 与体系中的水反应生成CH3OH 和副产物三氯乙酸,比传统脱水剂2-CP 产生的2-PA、吡啶甲酸甲酯等相比更容易回收,且以MTCL 为脱水剂DMC 收率能提高两倍以上。MTCL 价格较低、安全性更高,但是在CuCeO2上反应也导致副产物甲缩醛(DMM)和甲酸甲酯(MF)的生成,这可能与CH3OH 在CuO上的脱氢和缩醛化反应有关[86,87]。
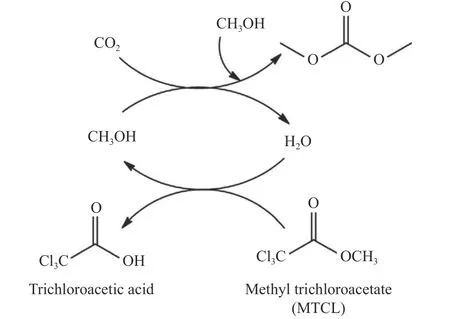
图25 MTCL 促进CO2 和CH3OH 合成DMC 反应机理[85]Figure 25 Reaction mechanism of synthesis of DMC from CO2 and CH3OH promoted by MTCL[85](with permission from Elsevier)
2022 年,张福灿等[88]报道了α-单取代炔丙醇、CO2和CH3OH 三组分耦合生成DMC 的新反应路径。详细考察催化剂、共催化剂、催化剂用量、溶剂、温度、原料比、压力和时间等因素对CH3OH 转化率和DMC 选择性的影响。研究表明,以磺胺嘧啶银为催化剂,超强碱1,8-二氮杂二环十一碳-7-烯(DBU)为共催化剂协同催化α-单取代炔丙醇、CO2和CH3OH 三组分耦合生成DMC。在最优条件下,DMC 选择性为89.5%,DMC 收率为55.6%。
3.3 反应器脱水工艺
2003 年,Li 等[89]以Cu-KF/MgSiO 为催化剂,在固定床膜分离反应器上考察三种负载型膜(SiO2无机膜、聚酰亚胺-SiO2杂化膜、聚酰亚胺-TiO2杂化膜)对CO2和CH3OH 合成DMC 的影响,固定床膜分离反应器构型如图26 所示。在反应温度130 ℃、CO2压力1.0 × 106Pa 和CH3OH∶CO2=2∶1(物质的量比)条件下,Cu-KF/MgSiO 催化剂上CH3OH 转化率和DMC 选择性分别为6.55%和90%。在上述相同条件下,与非负载型膜参与反应相比,三种负载型膜上DMC 收率分别提高0.02%、0.16%、0.07%。利用膜分离技术可以实现对水的分离,但存在设备相对复杂、膜透过率有限、反应压力对分离效果影响较大等问题。
沸石膜在气体或液体混合物分离方面具有巨大潜力。2015 年,Wang 等[90]制备出亲水性Linde Type A(LTA)和Sodalite(SOD)膜,由于SOD 膜孔径只有2.7 Å左右,允许动力学直径较小的H2O 分子(2.6 Å)通过,而CH3OH 分子(3.8 Å)被排除在外,从而选择性脱除体系中的水,促进膜反应器中CO2和CH3OH 合成DMC。室温下,LTA 膜 将H2O/CH3OH 混合物的渗透蒸发分离系数从2.8 提高到7.4。LTA 膜对H2O/DMC 混合物的分离性能也很好,离子交换前后选择性分别为800 和1000。此外,在125–200 ℃高温下,SOD 膜可以成功将水从CH3OH、DME 和DMC 中分离出来。
Hu 等[91]在gPROMS 平台设计出带有固定床反应器的均质RD(反应精馏)模型,包括用于环氧乙烷(EO)水合和轻重组分分离的精馏塔,以及用于合成DMC 的气相侧反应器,如图27 所示。CO2和CH3OH 进入精馏塔发生反应,从上塔流出未转化的CO2和CH3OH 以一定化学计量比引入侧反应器继续合成DMC,侧反应器产物流回精馏塔中;EO 和精馏塔的水结合生成乙二醇(EG),且只发生在塔底端液相中。在140 ℃和6.0 × 105Pa 条件下,该工艺将固定床反应器中约10%的CH3OH平衡转化率提高到99.5%,DMC 选择性为100%。另外,由于EO 水合作用的促进,再沸器和冷凝器的热负荷分别降低22%和1.6%。
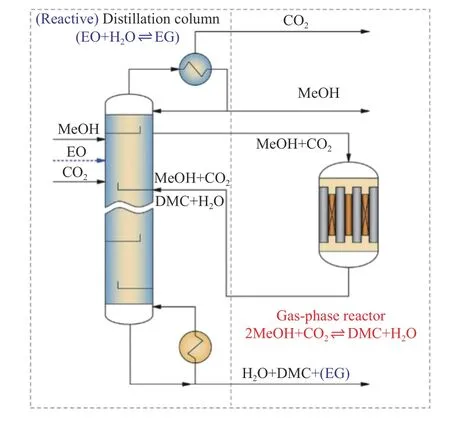
图27 CO2 和CH3OH 合成DMC 新型原位脱水反应器结构示意图[91]Figure 27 Schematic diagram of a novel in-situ dehydration reactor for CO2 and CH3OH to synthesize DMC[91](with permission from Elsevier)
Wu 等[92]利用Aspen Plus 开发出一种常规工艺和三种强化工艺,并对该工艺进行经济性和环保性评估。结果表明,与传统工艺相比,使用环氧乙烷(EO)水合精馏塔和气相侧反应器结合的强化工艺(RDSR-HI 和RDSRT 工艺,如图28)可以节省88%以上年总成本;此外,工艺流程中能源消耗导致的CO2排放量小于作为原料消耗的CO2量。
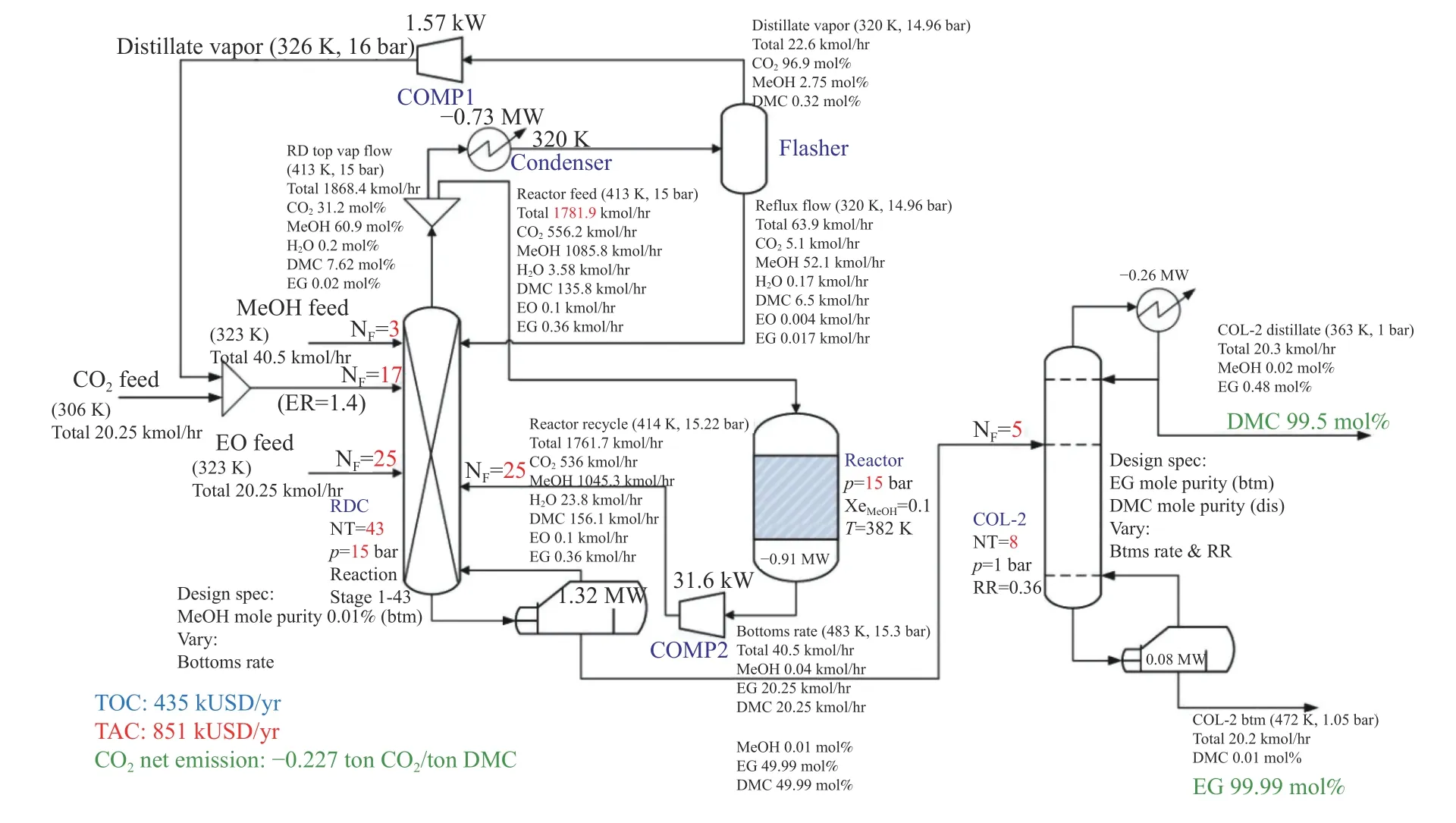
图28 侧反应器强化全脱水反应精馏工艺流程图[92]Figure 28 Process flow diagram of side reactor enhanced total dehydration reaction distillation[92](with permission from ACS Publication)
Zhou 等[93,94]采用水热法在α-Al2O3上成功制备出八面沸石型结构分子筛(FAU)膜,并将Ce0.8Zr0.2O2催化剂负载在FAU 分子筛膜上,在如图29 所示的固定床反应器中测试催化剂活性,在反应温度150 ℃和CO2压力5.0 × 105Pa 条件下,单位时间内DMC 收率为0.042 mmol/g。此外,在渗透汽化装置中,测试FAU 分子筛膜对10%H2O/90%DMC 混合溶液的分离性能,在80 ℃条件下,连续脱水9 h 后,进料侧DMC 浓度达到99%,FAU 膜表现出良好的分离性能。

图29 膜催化剂活性评价装置示意图[94]Figure 29 Schematic diagram of the equipment for membrane catalyst activity evaluation[94]
4 总结与展望
在过去几十年,由CO2和CH3OH 直接合成DMC 方法引起广泛关注,理论和实验结果均证明该路线可行,然而,合成DMC 低收率问题仍未得到根本解决。
通过各种耦合脱水体系可以打破CO2和CH3OH直接合成DMC 反应热力学平衡限制,在一定程度上提升DMC 收率。然而,现阶段无论是化学还是物理脱水方法都存在一些问题:多数化学脱水剂价格昂贵,在经济上不具备竞争优势;部分脱水剂毒性较高,安全性差,违背了CO2和CH3OH 合成DMC 反应低毒和绿色的初衷;化学耦合脱水方法对DMC 合成的促进作用未能达到理想程度,加上各种脱水剂所导致的难以控制的副反应,使产物分离难度和成本增加;膜分离技术存在设备相对复杂、膜透过率有限、DMC 收率低等问题;另外,一些新型反应器脱水工艺只处于开发模拟阶段。
目前,关于CO2和CH3OH 直接合成DMC 的速率控制步骤是CO2活化还是CH3OH 活化尚无定论。所以,确定催化剂上反应活性位点和速率控制步骤对于高活性催化剂以及高效脱水剂的精准设计和开发具有重要指导意义。此外,开发对水选择渗透性强的膜材料,构建和实施新型脱水工艺,如反应精馏、膜反应器与化学脱水剂耦合工艺等都是研究者们探索的方向。
