头孢美唑钠合成工艺的改进
2023-11-09李鹏
李鹏
(浙江东邦药业有限公司,浙江 临海 317016)
(6R,7R)7-甲氧基-7-氰甲基-硫乙酰胺-3-甲基-四唑-硫甲基头孢钠,英文名为 :(6R,7R)7-methoxy-7-cyano-methyl-thioacetamide-3-methyl-tetrazole-thiomethylcephalosporin sodium,英文名为 :Cefmeazole Sodium,分子式 :C15H16N7NaO5S3,结构如图1。
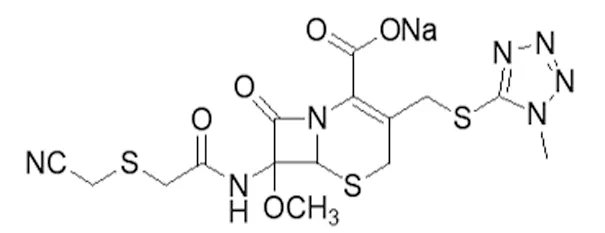
图1 (6R,7R)7-甲氧基-7-氰甲基-硫乙酰胺-3-甲基-四唑-硫甲基头孢钠分子结构
头孢美唑钠为β-内酰胺类抗生素、头孢菌素类药。适用于治疗由对头孢美唑钠敏感的金黄色葡萄球菌、大肠埃希菌、肺炎杆菌、变形杆菌属、摩氏摩根菌、普罗威登斯菌属、消化链球菌属、拟杆菌属、普雷沃菌属(双路普雷沃菌除外)所引起的下述感染:败血症,急性支气管炎、肺炎、肺脓肿、脓胸、慢性呼吸道疾病继发感染,膀胱炎、肾盂肾炎;腹膜炎;胆囊炎、胆管炎,前庭大腺炎、子宫内感染、子宫附件炎、子宫旁组织炎,颌骨周围蜂窝织炎、颌炎。与其他β-内酰胺类抗生素、头孢菌素类药相比,由于其母核7位α-H 被甲氧基所取代,从而增强了其母核的立体位阻,对β-内酰胺酶高度稳定,对产β-内酰胺酶以及不产β-内酰胺酶的敏感菌具有相同的抗菌活性[1]。头孢美唑钠由日本三共制药株式会社开发研制[2],1980年4月首次在日本上市,中国首次进口注册时间为1992年。目前,中国有多家厂家生产头孢美唑钠[3-6],但合成采用的路线复杂,所用试剂价格贵,对环境污染大,操作要求高;导致头孢美唑钠收率低,成本高,质量差,因此,对头孢美唑钠合成工艺进行改进具有很大的社会效益和经济效益。文献报道的头孢美唑钠的合成路线主要有2条:
(1)如图2路线1[7]以7-ACA为起始原料,经酯交换、上胺基保护、羧基硅酯化、氨基活化、甲氧化、二次羧基硅酯化和侧链缩合,得头孢美唑酸,头孢美唑酸再和碳酸氢钠成盐得头孢美唑钠。该路线所用氯仿水分要求很高(小于0.01%),不容易回收再利用,能耗及污染较大,不利于工业化生产。


图2 头孢美唑钠合成路线1
(2)如图3路线2[8],以7-ACA为起始原料,经酯交换、羧基硅烷化、上氨基保护、酯化、上甲氧基得到7-MAC,然后在五氯化磷、DMAC和吡啶的作用下,上酰胺保护基,在三氯化铝和苯甲醚的作用下水解得到头孢美唑酸,再和碳酸氢钠成盐得头孢美唑钠。该路线所用原料易得,路线简单,适合工业化生产。
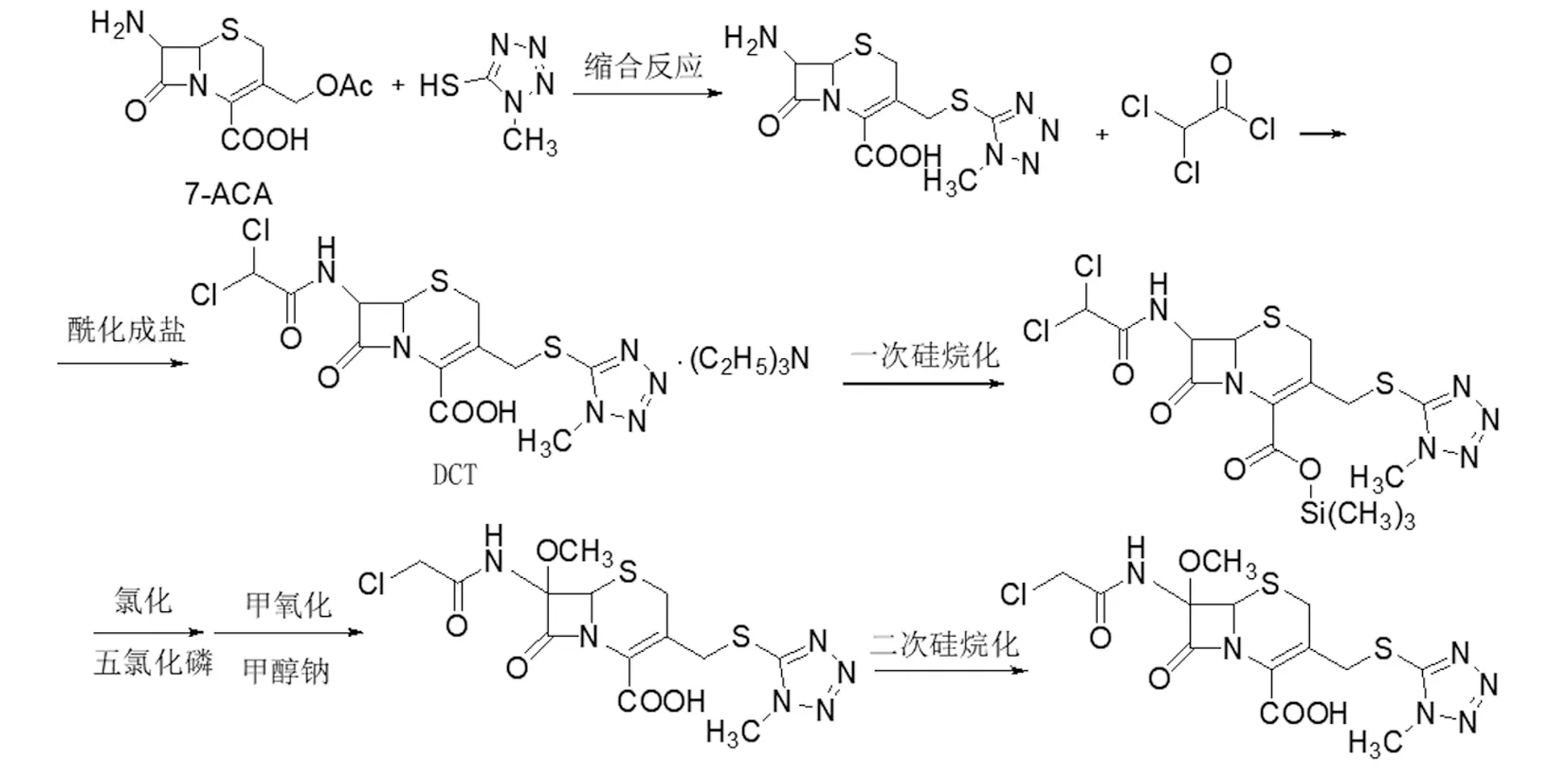

图3 头孢美唑钠合成路线2
参考路线2,对其合成路线进行改进,使得该合成路线更加简单、环保。路线2从7-MAC到头孢美唑钠的合成过程中,使用了五氯化磷、DMAC和吡啶等对环境不友好的试剂;为了解决这个问题,笔者用氯甲酸乙酯替代五氯化磷,氯甲酸乙酯和氰甲巯基乙酸钾反应生成混酐;混酐在三乙胺作用下和7-MAC反应生成化合物1。化合物1在三氯化铝和苯甲醚的作用下水解得到头孢美唑酸,再经过多次转相、结晶得高纯度头孢美唑酸;头孢美唑酸和碳酸氢钠成盐,冻干得头孢美唑钠。改进后的工艺不使用高污染试剂五氯化磷和吡啶,操作简单,降低了头孢美唑钠的原料成本,减轻了环境压力,有利于绿色生产;头孢美唑钠的色谱纯度提高。改进后的头孢美唑钠合成工艺见图4。
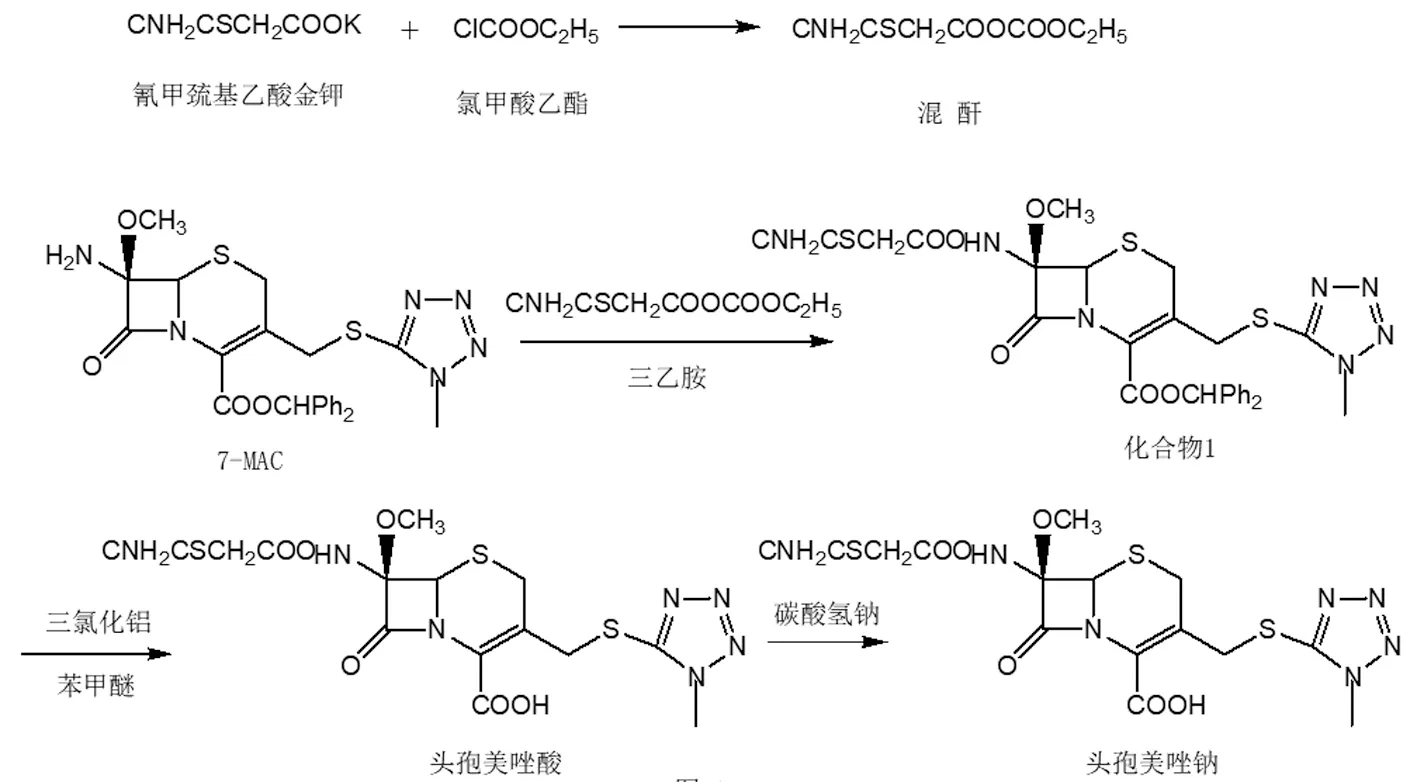
图4 改进后的头孢美唑钠合成工艺
1 实验部分
1.1 仪器和药品
Agilent1200/1260高效液相色谱仪,ARX-500型核磁共振仪,ALPHA傅里叶变换红外光谱谷仪,梅特勒托利多FE28-Standard 实验室台式pH计酸度计、真空干燥烘箱、过滤器、三口烧瓶等。7-MAC(浙江东邦药业有限公司),氰甲巯基乙酸钾(浙江东邦药业有限公司),为工业级。二氯甲烷、氯甲酸乙酯、三乙胺、甲醇、苯甲醚、三氯化铝、碳酸氢钠、767活性炭、甲苯、乙酸乙酯、精制盐酸、甲基异丁基酮,均为试剂级(国药集团)。
1.2 合成过程
1.2.1 化合物1的合成
在1 000 mL三口烧瓶中加入二氯甲烷500 mL,氰甲巯基乙酸钾(21.6 g,0.158 mol),降温至0~5 ℃,搅拌30 min,滴加氯甲酸乙酯(18.0 g,0.166 mol),30 min滴完,在0~5 ℃,搅拌2 h后降温至-30~-25 ℃,加入7-MAC(78.5 g,0.15 mol),搅拌5 min,滴加三乙胺(16.0 g,0.158 mol),60 min滴完,终点pH值在6.5~7.0,并在-30~-25 ℃保温反应2 h。 TLC点板(展开剂:甲苯/乙酸乙酯体积比3∶1)确认反应结束后,加水300 mL,控制温度5~10 ℃搅拌30 min,静止分层,水层用20 mL二氯甲烷萃取,合并二氯甲烷层;二氯甲烷层再加水200 mL,控制温度5~10 ℃搅拌30 min,静止分层,水层用20 mL二氯甲烷萃取,合并二氯甲烷层。减压蒸出二氯甲烷(内温控制小于20 ℃),蒸至有固体析出时,加甲醇400 mL,升温至20~25 ℃,搅拌30 min,降温至0 ℃,并搅拌1~2 h,过滤,滤饼用100 mL冰甲醇漂洗,滤饼在45~50 ℃真空干燥6 h,得 91.06 g化合物1,收率96%,色谱纯度98.5%。
1.2.2 头孢美唑酸的合成
在250 mL三口烧瓶中加入三氯化铝(20 g,0.15 mol),二氯甲烷100 mL,搅拌降温至0~5 ℃,滴加苯甲醚(27 g,0.25 mol),30 min滴完,继续搅拌30 min,备用。在另一个2 000 mL三口烧瓶中加入化合物1(60 g,0.094 mol),二氯甲烷500 mL 30~35 ℃搅拌1 h,溶清后降温至-25~-20 ℃,滴加三氯化铝苯甲醚溶液,控制温度-25~-20 ℃,30 min滴完,搅拌反应2 h。TLC点板(展开剂:甲苯/乙酸乙酯体积比3∶1)确认反应结束后,加丙酮200 mL,饮用水400 mL,精制盐酸22 g,20~25 ℃搅拌1 h,静止30 min,分层,水层用30 mL二氯甲烷萃取两次,合并有机层。向有机层中加饮用水200 mL,20~25 ℃搅拌15 min,静止30 min,分层,水层用30 mL二氯甲烷萃取,合并有机层。
向有机层加水100 mL,降温至0~5 ℃,滴加5%碳酸氢钠水溶液至pH值在6.0~6.5,搅拌30 min,静止30 min,分层,有机层用50 mL饮用水萃取,并合水层,向水层加767活性炭1 g,0~5 ℃,搅拌30 min,过滤,滤饼用20 mL饮用水漂洗,合并至滤液中,加入中性氧化铝1 g,0~5 ℃,搅拌30 min,过滤,滤饼用20 mL饮用水漂洗,合并至滤液中。
向滤液加乙酸乙酯500 mL,降温至0~5 ℃,滴加10%的精制盐酸水溶液,至pH值1.5~2.0,搅拌30 min,静止30 min,分层,水层用50 mL乙酸乙酯萃取,合并乙酸乙酯层。向乙酸乙酯层加入767活性炭1 g,0~5 ℃,搅拌30 min,过滤,滤饼用20 mL乙酸乙酯漂洗,合并漂洗液和滤液。向乙酸乙酯反应液中加纯化水100 mL,降温至0~5 ℃,滴加5%碳酸氢钠水溶液至pH值在6.0~6.5,搅拌30 min,静止30 min,分层,乙酸乙酯层用20 mL纯化水萃取,合并水层。
向水相加767活性炭1 g,0~5 ℃,搅拌30 min,过滤,滤饼用20 mL纯化水漂洗,合并至滤液中。向滤液中加乙酸乙酯30 mL,甲基异丁基酮50 mL,升温至20~25 ℃,滴加5%盐酸水溶液至pH值=3.0~3.5,加入头孢美唑酸晶种1 g,搅拌2~3 h,继续滴加5%盐酸水溶液,维持pH值=3.0~3.2,有大量白色固体析出,搅拌30 min,养晶结束后,滴加5%盐酸水溶液至pH值=1.5~2.0,搅拌30 min,降温至0~5 ℃,保温1 h后,抽滤,滤饼分别用纯化水200 mL、甲基异丁基酮 50mL、二氯甲烷 50 mL洗涤,得头孢美唑酸湿品;头孢美唑酸湿品在40~45 ℃减压干燥得白色固体40.1 g,收率90.5%。色谱纯度99.7%。熔点125~126 ℃,质量分数90%。1H NMR(400 MHz,DMSO)δ:3.45(1H,d),3.84(1H,d),3.50(1H,s),3.63(3H,s),3.70(2H,s),3.95(3H,s),4.20(1H,d),4.40(1H,d),5.08(1H,s)。IR(KBr)3 431,3 398,2 974,2 955,2 918,2 250,1 765,1 694,1 635,1 507,1 483 cm-1。
1.2.3 头孢美唑钠的合成
在500 mL三口烧瓶中加入头孢美唑酸(40 g,0.042 mol),纯化水200 mL,降温至0~5 ℃,分批次加碳酸氢钠(3.56 g,0.042 mol),30 min加完,至pH值=5.5~6.0,头孢美唑酸溶清后,吹氮气30 min,过滤,滤液在冻干机中冻干得头孢美唑钠白色固体41.2 g,收率98.6%,色谱纯度99.0%。
2 分析与讨论
2.1 化合物1的合成
氰甲巯基乙酸钾和氯甲酸乙酯反应生成混酐(活化剂);混酐在三乙胺作用下和7-MAC反应生成化合物1。制备化合物1时,利用氯甲酸乙酯替代文献工艺的五氯化磷,三乙胺替代吡啶和DMAC。优化后的工艺减少了试剂使用的种类;氯甲酸乙酯替代五氯化磷,提高了反应速度,五氯化磷在二氯甲烷中溶解度小,需要加入DMAC增加溶解度,在活化氰甲巯基乙酸钾时,需要10 h以上,但氯甲酸乙酯为液体和二氯甲烷互溶,活化氰甲巯基乙酸钾只需要2 h,缩短了反应时间,而减少了废水含磷产物(磷在废水中很难去除);三乙胺替代吡啶,降低了原料成本(吡啶市场价格比三乙胺贵),减少现场恶臭(吡啶在工业化生产过程中属于恶臭类物料,在国内化工园区入园时一般属于限制入园类物料),三乙胺比吡啶更易回收,而能重复使用,减少了废液量的产生。
在合成化合物1时,滴加三乙胺的反应温度、滴加速度和使用量对化合物的收率和质量有影响;反应温度高,滴加三乙胺速度过快,反应结束三乙胺过量,易产生异构体;经过大量试验,确定了滴加三乙胺的最佳温度为-30~-25 ℃,滴加时间为60 min,三乙胺的最佳用量为终点pH值在6.5~7.0。优化后的化合物1合成工艺,试剂种类减少,减少了废水中含磷试剂的排放,降低了原料成本,缩短了反应时间,操作更简单。
2.2 头孢美唑酸的合成
合成头孢美唑酸时,在低温条件下,利用三氯化铝和苯甲醚脱羧基保护;并经过多次转相提高了头孢美唑酸的质量(色谱纯度达到99.7%,头孢美唑酸的溶液颜色达到了黄色1号)。头孢美唑酸到头孢美唑钠,没有去杂质功能,头孢美唑酸的质量直接决定了头孢美唑钠的质量,头孢美唑酸转相过程的温度控制及pH值控制非常关键,影响头孢美唑酸的颜色和色谱纯度。经试验确认,转相最佳温度为0~5 ℃,温度过高,头孢美唑酸的颜色变深。头孢美唑酸由有机相转水相pH值控制5.5~6.0,pH值过高,反应液颜色变深,头孢美唑酸杂质变大;pH值过低,转相不够充分,头孢美唑酸收率低。加晶种时反应液pH值会影响头孢美唑酸的结晶,加晶种最佳pH值为3.0~3.5,pH值过高,加晶种后全部溶解,晶种没有起到作用,pH值过低,容易爆析,头孢美唑酸颜色差,杂质大。优化后的工艺,提高了头孢美唑酸的质量。
2.3 头孢美唑钠的合成
合成头孢美唑钠时,头孢美唑酸和碳酸氢钠的反应温度对孢美唑钠的质量有影响,温度过高,头孢美唑钠降解产生四氮唑和内酯两个杂质。头孢美唑钠质量标准里面有pH值要求,为了达到头孢美唑钠质量要求,头孢美唑酸和碳酸氢钠的反应终点pH值很关键,经大量试验确认,最佳pH值为5.5~6.0。
3 结论
以7-MAC为原料,在氰甲巯基乙酸钾、氯甲酸乙酯和三乙胺的作用下生成化合物1;化合物1在三氯化铝和苯甲醚的作用下,并经多次转化,结晶得到头孢美唑酸;头孢美唑酸和碳酸氢钠成盐,冻干得到头孢美唑钠。本路线操作简单,制备的头孢美唑钠质量好,所用原料便宜而对环境污染小,节约成本,设备要求不高,缩短了反应时间,适合工业化生产。
