固相萃取-高效液相色谱-串联质谱法同时测定鸭蛋中4种硝基咪唑类药物残留
2023-11-08王圆圆贾浩程户瑞刚曹玲芝
王圆圆,李 宁,贾浩程,户瑞刚,曹玲芝,肖 琎
(1.唐山市农业综合行政执法支队,河北唐山 063000;2.遵化市农业农村局,河北遵化 064200;3.唐山市食品药品综合检验检测中心,河北唐山 063000;4.唐山市动物检疫站,河北唐山 063000;5.衡水市农业综合行政执法支队,河北衡水 053000)
我国是世界上最大的水禽生产国,其中鸭饲养量占我国水禽饲养总量的70%以上,鸭蛋消费量占禽蛋总消费量的30%以上[1]。在绿色发展理念的倡导下,蛋鸭养殖方式逐步从传统水养转为离水旱养[2]。为减少养殖过程中疾病的发生,养殖者通常使用抗生素进行预防及治疗。但是目前抗生素滥用、误用以及不按休药期使用等现象频发,抗生素残留超标已成为鸭蛋中药物残留的主要指标,严重影响着人们的饮食安全[3-5]。
硝基咪唑类药物具有致突变性和潜在的致癌性,被许多国家列为违禁药物[6]。在我国《食品安全国家标准 食品中兽药最大残留限量》(GB 31650—2019)中,甲硝唑、地美硝唑被列为不得在动物性食品中检出的兽药[7-8]。
甲硝唑、地美硝唑及其代谢标志物残留的检测方法主要有气相色谱法、液相色谱法、酶联免疫吸附法、液相色谱-串联质谱法等。液相色谱-串联质谱法具有特异性强、灵敏度高等优点,已成为目前检测甲硝唑、地美硝唑及其代谢物残留的首选方法[9-11]。本研究采用同位素内标法定量方式,通过校正基质效应,提高了分析准确性,为鸭蛋中甲硝唑、地美硝唑及其代谢物残留测定提供了技术支撑。
1 材料与方法
1.1 仪器、试剂与材料
高效液相色谱仪、三重四极杆联用质谱仪(5 500+),均为AB SCIEX公司产品;MEP固相萃取柱3 CC,为上海安谱实验科技股份有限公司产品;Oasis MCX 固相萃取柱3 CC(60 mg),为美国Waters公司产品;Syncore Plus多样品平行蒸发定量浓缩仪,为瑞士步奇有限公司产品;多管涡旋振荡器,为德国Heidoph公司产品;氮吹仪,为杭州聚同电子有限公司产品;高功率超声波清洗器,为昆山市超声波仪器有限公司产品;低温冷冻离心机,为美国赛默飞科技有限公司产品。
甲硝唑、甲硝唑-D3、羟基甲硝唑、羟基甲硝唑-D2、地美硝唑、二甲硝咪唑-D3、羟基地美硝唑、羟甲基甲硝咪唑-D3等标准物质,均购自天津阿尔塔科技有限公司;乙腈、异丙醇(均为色谱纯)以及0.45、0.22 μm滤膜,均购自美国Dikma公司;五氟苯基色谱柱100A(2.6 μm,100 mm×2.1 mm),为美国Phenomenex公司产品。柠檬酸缓冲溶液配制:取21 g一水合柠檬酸和20 g六水合氯化镁溶于900 mL水中,以氨水调节pH为2.5,最后用水定容至1 L。
1.2 标准溶液配制
分别将甲硝唑、地美硝唑、羟基甲硝唑、羟基地美硝唑和对应的4种同位素内标标准品(质量浓度均为100 μg/mL),用甲醇稀释定容为标准物质中间液(10 μg/mL),-20 ℃保存。
分别准确移取甲硝唑、地美硝唑、羟基甲硝唑、羟基地美硝唑的标准物质中间液以及4种内标标准物质中间液,用甲醇-水(v:v= 1:1)稀释,配制成质量浓度为100 ng/mL的混合标准工作液以及混合内标标准工作液,4 ℃保存。
1.3 标准曲线绘制
将8份鸭蛋空白样品,经过前处理制成空白基质溶液。用鸭蛋空白基质溶液将甲硝唑等4种混合物质标准工作液配制质量浓度为0.5、1.0、2.0、4.0、10.0、50.0、100.0、200.0 μg/L的系列溶液,4种对应混合内标标准工作液质量浓度为5.0 μg/L,将8种浓度的标准物质工作液分别上机后,绘制标准曲线。
1.4 样品前处理
准确称取样品5 g(精确至0.01 g)放入50 mL离心管中,加入混合内标标准工作液50 μL;依次加入2.5 mL柠檬酸缓冲液、15 mL乙腈溶液,4 000 r/min涡旋混合提取5 min;5 000 r/min离心5 min,取上清液至平行蒸发蒸馏瓶中,按照上述步骤重复提取并附加超声波提取10 min;合并提取液,加入3 mL异丙醇,(40 ± 1)℃蒸发浓缩至5 mL以下,将蒸发后的浓缩液转移至新离心管;用柠檬酸缓冲液(10 mL)润洗蒸馏瓶,8 000 r/min低温离心10 min,以0.45 μm滤膜过滤,待净化。
将MEP柱和MCX固相萃取柱按照自上而下顺序安装好,依次用2.5 mL甲醇、2.5 mL水、2.5 mL柠檬酸缓冲液(pH 2.5)活化,将全部净化液通过净化柱;依次用3 mL柠檬酸缓冲液(pH 2.5)、3 mL水、3 mL甲醇-水(v:v= 1:19)淋洗固相萃取柱;去掉MEP柱,依次用2 mL甲醇、4 mL氨水-甲醇(v:v= 1:19)洗脱MCX固相萃取柱的待分析成分;收集洗脱液,在45 ℃条件下以氮气吹至干;用甲醇-0.1%甲酸水(v:v=1:10)溶液溶解残渣,以0.22 μm滤膜过滤后供液相色谱-串联质谱测定。
1.5 液相色谱条件
色谱柱:五氟苯基色谱柱;进样体积3 μL,流速0.4 mL/min,柱温40 ℃;流动相溶剂A为0.1%甲酸水,溶剂B为甲醇。梯度洗脱程序见表1。
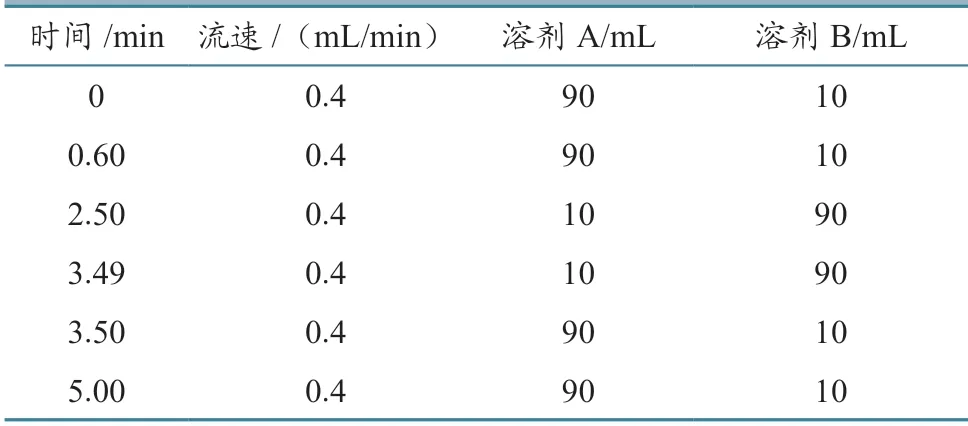
表1 梯度洗脱程序
1.6 质谱条件
电喷雾正离子扫描模式:以多反应监测(MRM)模式扫描,电喷雾电压5 500 V,离子源温度500 ℃,气帘气压力35 psi,辅助气压力50 psi。4种硝基咪唑类药物定量、定性离子对信息见表2。
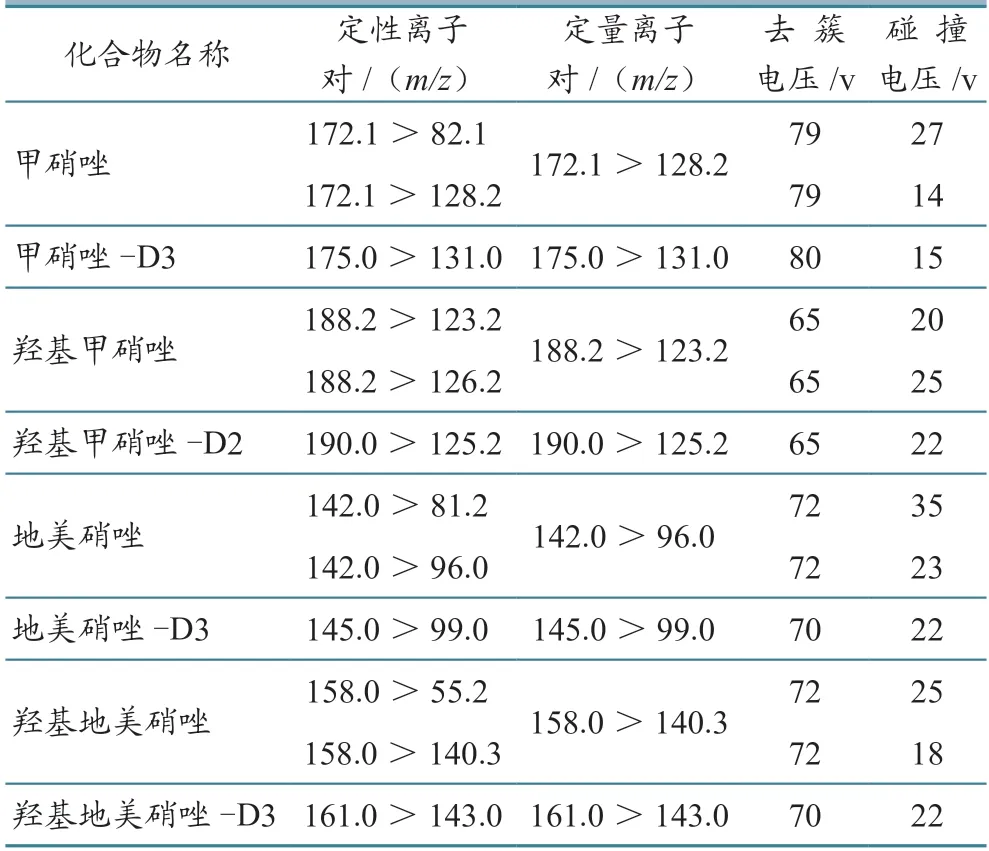
表2 4种硝基咪唑类药物质谱分析参数
2 结果与讨论
2.1 提取溶剂选择
本试验分别将乙酸乙酯、乙腈、甲醇等3种有机试剂与柠檬酸缓冲液(pH 2.5)进行搭配组合提取,并分别比较了鸭蛋中加标回收率。结果表明:乙腈+柠檬酸缓冲液(pH 2.5)混合提取的加标回收率最高,4种待测物质的加标回收率均在90%~110%范围内。鸭蛋富含蛋白质、磷脂等成分,在提取过程中,先加入柠檬酸缓冲液,利用盐析作用使鸭蛋中蛋白质缓慢沉淀,酸性条件下更有利于提高4种硝基咪唑类药物的回收效率。加入异丙醇可以在平行蒸发过程中降低溶液中气泡的产生,减少待测物损失。经综合考虑,采用柠檬酸缓冲液(pH 2.5)+乙腈作为提取溶剂,其效果最好。
2.2 提取方式优化
本试验对使用提取液提取1次与2次的效果进行了比较验证,发现4种硝基咪唑类药物的加标回收率均为90%~110%,但是两种提取方式的结果偏差分别为12.2%~15.3%和4.5%~8.7%,所以提取2次的方式结果更为稳定,准确性更好。本试验还比较了涡旋震荡提取和超声波助提对结果的影响,发现超声波助提有利于提升回收结果的稳定性,偏差相对较小,超声波提取1次与2次对回收结果基本无影响。此外,分别比较了超声5、10、15 min的回收效果,结果发现超声10 min的效果最好。提取方式采用涡旋+超声的处理方式,简单方便且回收效果良好。因此,本试验采用混合溶剂提取2次,并在第2次采用超声波助提10 min的方式。
2.3 固相萃取柱选择
本试验比较了ProElut SCX(Dikma公司)、CNW Poly-Sery MCX SPE(上海安谱实验科技股份有限公司)、Oasis MCX(Waters公司)3种混合阳离子固相萃取柱(规格均为3CC,60 mg)对回收效果的影响。结果显示,甲硝唑、地美硝唑、羟基甲硝唑、羟基地美硝唑在10 ng/kg添加水平下,ProElut SCX固相萃取柱的平均回收率分别为70.3%、67.4%、73.3%、71.2%;CNW Poly-Sery MCX SPE固相萃取柱的平均回收率分别为85.3%、83.8%、79.5%、80.4%,Oasis MCX固相萃取柱的平均回收率分别为98.2%、95.3%、94.9%、92.7%。综上,本试验优选Oasis MCX固相萃取柱。试验表明,不同厂家的固相萃取柱在回收效果上存在差异,这可能与固相萃取柱的填料、交换基团等存在差异有关。固相萃取技术种类繁多,因此选择合适的固相萃取柱显得尤为重要。
2.4 基质效应
为验证本试验方法去除基质干扰的效果,在空白鸭蛋样品中添加10 ng/kg的混合标准工作液,根据相同浓度的纯标准溶液的响应强度与空白添加样品提取后的响应强度的比值计算基质效应(ME)。经计算,鸭蛋中4种硝基咪唑类药物的ME为0.88~1.09,提示ME不明显。ME主要分为基质增强效应和基质减弱效应,均影响分析结果的准确性。为减少ME的干扰,本研究优化前处理,提高净化效果,引入内标校正,提高了结果的准确性。
2.5 液相色谱与质谱条件优化
本试验分别将甲醇、乙腈作为有机相,并与作为水相的纯水和0.1%甲酸-水进行混合搭配验证,比较等度洗脱与梯度洗脱时4种硝基咪唑类药物的相应度及分离度。结果显示:梯度洗脱时,甲醇作为有机相,0.1%甲酸-水作为水相,4种硝基咪唑类药物的响应值及峰形较好。本试验还分别比较验证了BEH C18、HSS T3、Hypersil GOL、Kinetex F5等色谱柱的响应值及色谱峰形,发现4种色谱柱在保留时间和响应值上有差异。综合考虑发现,五氟苯基色谱柱(Kinetex F5)100A(2.6 um,100 mm×2.1 mm)的响应值及峰形最佳(图1)。不同规格的色谱柱分离效果不同,尤其是保留时间、响应值的差异明显。因此在方法开发时,可以尝试使用不同规格的色谱柱,选择效果最佳的。

图1 4种硝基咪唑类药物(4 μg/L)及其内标物离子色谱图
2.6 线性关系及检出限、定量限
将4种硝基咪唑类药物标准工作液用鸭蛋空白基质溶液,分别配制成不同质量浓度的基质混合标准溶液,内标质量浓度为5 μg/L,经质谱检测绘制标准曲线。结果显示,4种硝基咪唑类药物在0.5~200.0 μg/L范围内线性关系良好,相关系数(R2)均大于0.997。以3倍基线噪声确定检出限,10倍基线噪声确定定量限,4种硝基咪唑类药物检出限为0.21~0.29 μg/kg,定量限为0.70~0.96 μg/kg(表3)。灵敏度的高低一般用检出限和定量限表示,二者值越小,表明方法灵敏度越高。结果表明,本试验建立的检测方法灵敏度高。
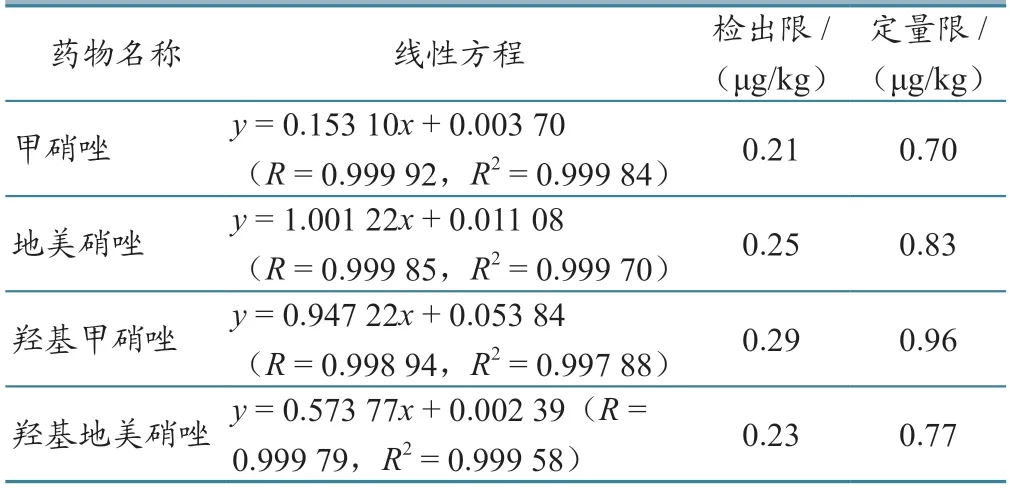
表3 4种硝基咪唑类药物的线性方程、检出限和定量限
2.7 精密度与加标回收率
选取鸭蛋空白样品,添加混合标准溶液(10 μg/kg),加入内标混合标准工作液50 μL,进行加标回收试验(n= 6),以基质内标法定量。结果(表4)显示,4种硝基咪唑类药物的加标回收率为90%~110%,相对标准偏差(RSDs)为4.76%~7.17%。精密度是指在同样试验条件下,反复测量相同基质样品所得测量值之间的接近程度。一般来说,回收率越高,相对标准偏差越小,表明此方法越可靠。本研究数据表明,所建立的方法可靠。
2.8 实际样品分析
采用该方法对农贸市场售卖鸭蛋中的4种硝基咪唑类药物残留情况进行检测,共计检测56份样品,结果检出甲硝唑药物残留样品1份,定量结果为1.05 μg/kg,其他3种药物均未被检出,表明硝基咪唑类药物在动物源性食品中添加的非法行为依然存在。
3 结论
本试验建立了鸭蛋中4种硝基咪唑类药物残留的检测方法,以内标法定量,结果更为准确,显著提高了分析的灵敏度、精密度。该方法回收率高、准确性好,完全满足4种硝基咪唑类药物残留的定性、定量需求,可为食品中污染物监测提供有效的技术支撑。
