重组牛疱疹病毒1型转移载体的构建与初步应用
2023-11-05戴莎莎马小静王景松田兴苗王健霖李继东
戴莎莎,马小静,王景松,田兴苗,王健霖,李继东
(宁夏大学 动物科技学院,宁夏 银川,750021)
关键字:牛疱疹病毒1型;同源臂;转移载体;同源重组
牛传染性鼻气管炎(infectious bovine rhinotracheitis, IBR)是由牛疱疹病毒1型(Bovineherpesvirus1, BHV-1)引起的一种接触性传染病,主要引起牛高热、呼吸困难和上呼吸道炎症等症状[1]。IBR的传染源主要为带毒病畜,可通过飞沫、交配、接触传播和吸血昆虫传播,病畜在自然条件下可不定期向外界排毒。牛是BHV-1自然感染的唯一宿主,以肉牛最为易感[2],奶牛次之。由于缺乏有效的治疗方法和抗病毒药物,接种疫苗仍是防止BHV-1传播流行最有效的方法[3]。
传统的灭活疫苗和弱毒疫苗存在免疫原性低或者减毒不充分等缺点,而基因工程疫苗弥补了这部分缺点。疱疹病毒可以作为病毒载体插入多个外源基因序列,诱导动物产生广泛的免疫反应,不仅可以诱导体液免疫和细胞免疫,还可以诱导特异性的黏膜免疫应答[4]。王子书等[5]利用细菌人工染色体(bacterial artificial chromosome, BAC)技术和病毒拯救技术在火鸡疱疹病毒(Herpesvirusofturkey, HVT)基因组中插入鸡传染性法氏囊病病毒(Infectiousbursaldiseasevirus, IBDV)的VP2基因,以及新城疫病毒(Newcastlediseasevirus, ND)的F基因,获得了共表达NDV和IBDV保护性抗原基因的重组HVT;Feng等[6]将非洲猪瘟病毒(Africanswinefevervirus, ASFV)的CD2V基因插入到TK、gE、gI基因缺失的伪狂犬病病毒(Pseudorabiesvirus, PRV)基因组中,为ASF疫苗研发奠定了基础;Shao等[7]将NDV的F基因插入到缺失US9基因的鸡传染性喉气管炎病毒(Infectiouslaryngotracheitisvirus, ILTV)基因组中,得到重组毒ILTV-ΔUS9-F,且重组病毒的生长特性与亲本病毒相似;Aligholipour Farzani等[8]构建了表达重组牛疱疹病毒4型N蛋白的质粒,并通过小鼠致死性攻毒试验验证了其对BALB/c小鼠的免疫原性和保护潜力。
经同源重组产生重组病毒需要构建一个转移载体,大部分转移载体只能用于一次特定的病毒重组。为了便捷地进行BHV-1的同源重组,笔者拟设计、构建一个能用于BHV-1重组的转移载体,采用双向CMV启动子分别控制标记基因GFP(绿色荧光蛋白基因)和外源抗原基因的表达,引入多克隆位点便于选择插入不同同源臂作为重组位点。构建转移载体将方便BHV-1的重组,并以此研究基因功能和研发重组病毒活载体疫苗,为IBR的防控提供技术支持。
1 材料与方法
1.1 毒株菌株与细胞
BHV-1 HVRI-002病毒株、BVDV NADL病毒株和牛肾(Madin-Darby bovine kidney, MDBK)细胞均由宁夏大学农学院预防兽医实验室保存,DH5α感受态细胞购自天根生化科技(北京)有限公司。
1.2 主要试剂
DNA/RNA提取试剂盒为Omega公司产品;反转录试剂盒购自南京诺唯赞生物科技股份有限公司;PrimeSTAR HS DNA Polymerase、PrimeSTAR HS DNA Polymerase with GC Buffer、DNA Marker DL 2000、6×loading buffer购自宝日医生物技术(北京)有限公司;DNA凝胶纯化回收试剂盒、质粒小提试剂盒购自天根生化科技(北京)有限公司;限制性内切酶、T4 DNA连接酶为NEB公司产品;氨苄青霉素钠购自北京博奥拓达科技有限公司;LB培养基购自青岛高科技工业园海博生物技术有限公司;Opti-MEM培养基、Lipofectamine 3000转染试剂购自赛默飞世尔科技公司;pVAX1质粒为Invitrogen公司产品,pIRES2-AcGFP1质粒为Clontech公司产品,由宁夏大学农学院预防兽医实验室保存;兔抗BVDV E2抗体购自北京博奥森生物技术有限公司;罗丹明标记山羊抗兔IgG抗体购自北京中杉金桥生物技术有限公司。
1.3 BHV-1转移载体prPgP的构建
在pVAX1质粒序列第753位碱基处设计上游引物,第695位碱基处反向设计下游引物,引物中引入酶切位点,用NsiⅠ酶切后构建质粒pVAX1-1,缺失部分多克隆位点;以pVAX1质粒131~735位互补序列为模板设计上、下游引物,引物中引入酶切位点,扩增CMV启动子和部分酶切位点的互补序列rCMV;以质粒pVAX1-1为模板,在第1位碱基处设计上游引物,在2 960位碱基处反向设计下游引物,引物中引入酶切位点,PCR扩增目的片段pVAX1-2;rCMV和pVAX1-2用SalⅠ、XmaⅠ双酶切后连接,构建重组质粒prPP;以pIRES2-AcGFP1质粒为模板,扩增GFP基因,用AflⅡ、NheⅠ双酶切后连接于prPP,得到重组质粒prPgP(图1)。利用Prime premier 5.0软件设计引物,序列见表1,由生工生物工程(上海)股份有限公司合成。以pVAX1质粒为模板扩增pVAX1-1、rCMV,以pVAX1-1为模板扩增pVAX1-2。以pIRES2-AcGFP1质粒为模板扩增GFP。PCR退火温度见表1。

表1 prPgP载体构建引物序列与酶切位点
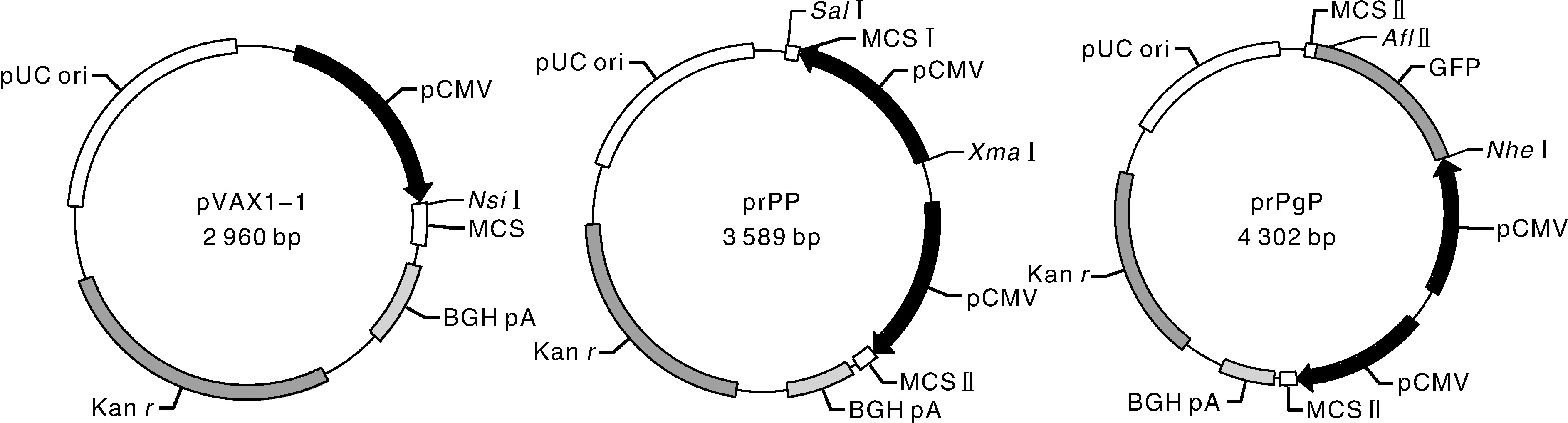
prPP 多克隆位点(MCS)Ⅰ:SalⅠ, NsiⅠ, BglⅡ, EcoRⅠ, PstⅠ, EcoR V, NotⅠ, XhoⅠ, XbaⅠ, ApaⅠ; prPP 多克隆位点(MCS)Ⅱ:ClaⅠ, SaIⅠ, BspEⅠ, BamHⅠ, KpnⅠ, HindⅢ, AflⅡ, PmeⅠ, NheⅠ, XmaⅠ。prPP multiple cloning site (MCS) Ⅰ: SalⅠ, NsiⅠ, BglⅡ, EcoRⅠ, PstⅠ, EcoR V, NotⅠ, XhoⅠ, XbaⅠ, ApaⅠ; prPP multiple cloning site (MCS) Ⅱ: ClaⅠ, SaIⅠ, BspEⅠ, BamHⅠ, KpnⅠ, HindⅢ, AflⅡ, PmeⅠ, NheⅠ, XmaⅠ.图1 载体prPgP构建示意图Fig.1 Schematic diagram of construction of universal vector prPgP
1.4 BHV-1 p△gErPgP-E2载体的构建
1.4.1 引物设计与目的片段扩增
利用Prime premier 5.0软件,根据BHV-1全基因组(AJ004801.1)、BVDVE2基因序列(M31182.1)设计引物,序列见表2,由生工生物工程(上海)股份有限公司合成。取BHV-1和BVDV病毒液,用Omega基因组提取试剂盒提取病毒基因组。对BVDV基因组进行反转录获得cDNA。以BHV-1 DNA为模板扩增重组同源臂gEL、gER序列,以BVDV cDNA为模板扩增E2基因。PCR退火温度见表2。用DNA凝胶纯化回收试剂盒回收PCR目的产物,储存于-20 ℃。

表2 p△gErPgP-E2质粒引物序列与酶切位点
1.4.2 p△gErPgP-E2重组质粒构建
用BamHⅠ、HindⅢ酶切gEL片段,用PstⅠ、EcoRⅤ酶切gER片段,依次将同源臂gEL和gER插入prPgP的多克隆位点Ⅰ(multiple cloning site, MCSⅠ)和MCSⅡ中;用BglⅡ、PstⅠ酶切BVDVE2基因,将E2连接到正向CMV启动子下的MCSⅡ中。重组质粒图谱见图2。重组质粒进行双酶切鉴定,并送生工生物工程(上海)股份有限公司测序。
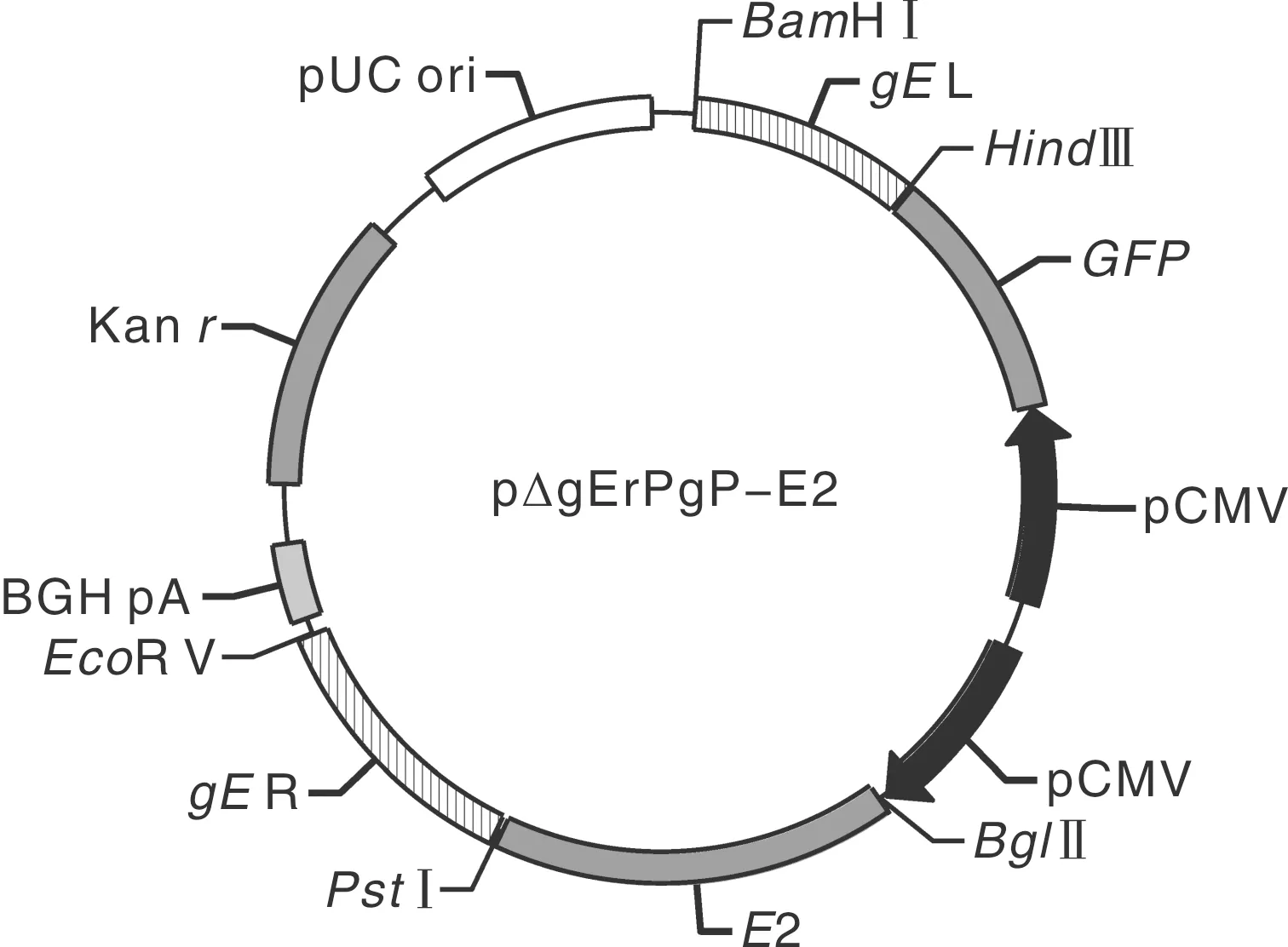
图2 重组载体p△gErPgP-E2构建示意图Fig.2 Schematic diagram of construction of recombinant vector p△gErPgP-E2
1.5 BHV-1重组病毒的产生与鉴定
1.5.1 细胞转染
将MDBK细胞铺于6孔细胞培养板,待细胞长满底部70%~90%时弃去培养基,用磷酸缓冲盐液(PBS)洗涤后加入500 μL BHV-1病毒液,37 ℃恒温吸附2 h后弃去病毒液,再次用PBS洗涤后加入2 mL Opti-MEM无血清培养基。p△gErPgP-E2质粒质量浓度为440 ng·μL-1,转染质粒量为2 500 ng。转染步骤如下:
(1)稀释Lipofectamine 3000转染试剂:125 μL opti-MEM和3.75 μL Lipofectamine 3000混合;(2)稀释质粒:将5.7 μL质粒加入125 μL opti-MEM中,再加入5 μL P3000混合均匀;(3)将稀释的转染试剂和质粒混匀,室温静置10~15 min;(4)向6孔板中加入质粒-脂质体复合物,37 ℃孵育6 h后换成含2% 胎牛血清(fetal bovine serum, FBS)的Dulbecco’s改良培养基(Dulbecco’s modified eagle medium,DMEM),继续培养2~4 d。当细胞病变达80%左右时收集细胞和培养液,于-80 ℃保存。
1.5.2 重组病毒纯化
将收获的细胞和培养液反复冻融3次,按10-1~10-4梯度进行稀释,接种至长满单层MDBK细胞的96孔板中,37 ℃培养2 h后换为含2% FBS的DMEM维持培养基,48 h后在倒置荧光显微镜下观察,收集荧光较多的细胞孔连续进行稀释、接种、培养,直到整个细胞孔全部呈现荧光。获得的重组病毒命名为rBHV-1-△gE/E2。
1.5.3 重组病毒PCR鉴定
收集重组病毒培养液,提取基因组DNA,分别设计针对缺失的gE基因、标记基因GFP、外源基因E2的引物,进行PCR以鉴定重组病毒的特征(表3)。
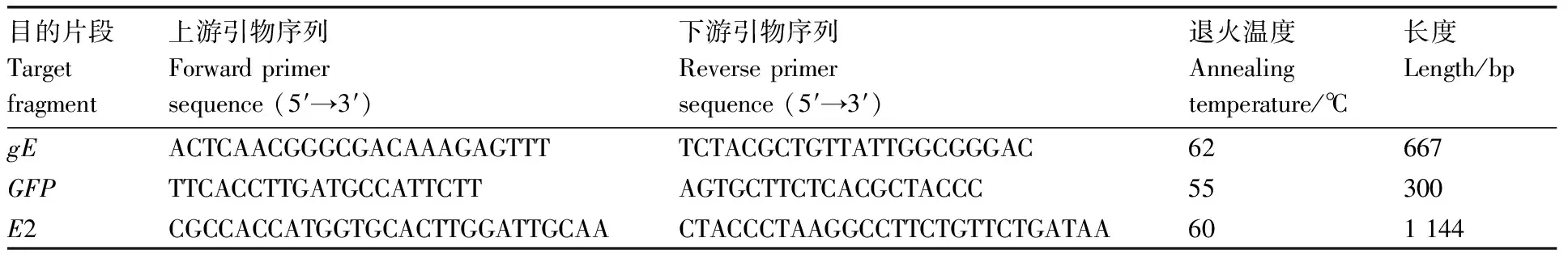
表3 重组病毒PCR鉴定引物序列
1.5.4 重组病毒与野毒株TCID50测定
分别将亲本病毒和重组病毒倍比稀释,设置10-1~10-10共10个稀释梯度,接种于长满单层MDBK细胞的96孔板中,于24、48、72 h后分别观察细胞病变情况。用Reed-Muench法计算病毒TCID50。
1.5.5 重组病毒体外生长曲线测定
用24孔板培养MDBK细胞,接种重组病毒。于12、24、36、48、60、72 h收集细胞和培养液,分别测定其TCID50,绘制病毒生长曲线。
1.5.6 间接免疫荧光试验(IFA)
将MDBK细胞铺于6孔板中,待细胞长满底部70%~90%时弃去培养基,用PBS洗涤后加入500 μL rBHV-1-△gE/E2重组病毒液和BHV-1亲本病毒液,37 ℃吸附2 h后弃去病毒液,换成含2%胎牛血清的DMEM,37 ℃培养24 h后取出,进行以下IFA试验。
(1)用PBS洗涤细胞后加入1 mL 4%多聚甲醛,37 ℃固定30 min,用PBS洗涤3次。(2)加入1 mL 1% Triton-100溶液,37 ℃透膜20 min,用PBS洗涤3次;(3)加入1 mL 2% 牛血清白蛋白(bovine serum albumin, BSA)溶液,37 ℃封闭1 h,用PBS洗涤3次;(4)加入1 mL 1∶400倍稀释的兔抗BVDV E2,4 ℃孵育过夜,用PBS洗涤3次;(5)加入1 mL 1∶50倍稀释的罗丹明标记的山羊抗兔IgG,37 ℃避光孵育1 h,用PBS洗涤3次,于倒置显微镜下观察结果并拍照。
2 结果与分析
2.1 prPgP载体目的片段PCR结果
PCR扩增出的pVAX1-1、rCMV、pVAX1-2和GFP预期大小分别是2 966、629、2 984、738 bp,结果符合预期(图3)。
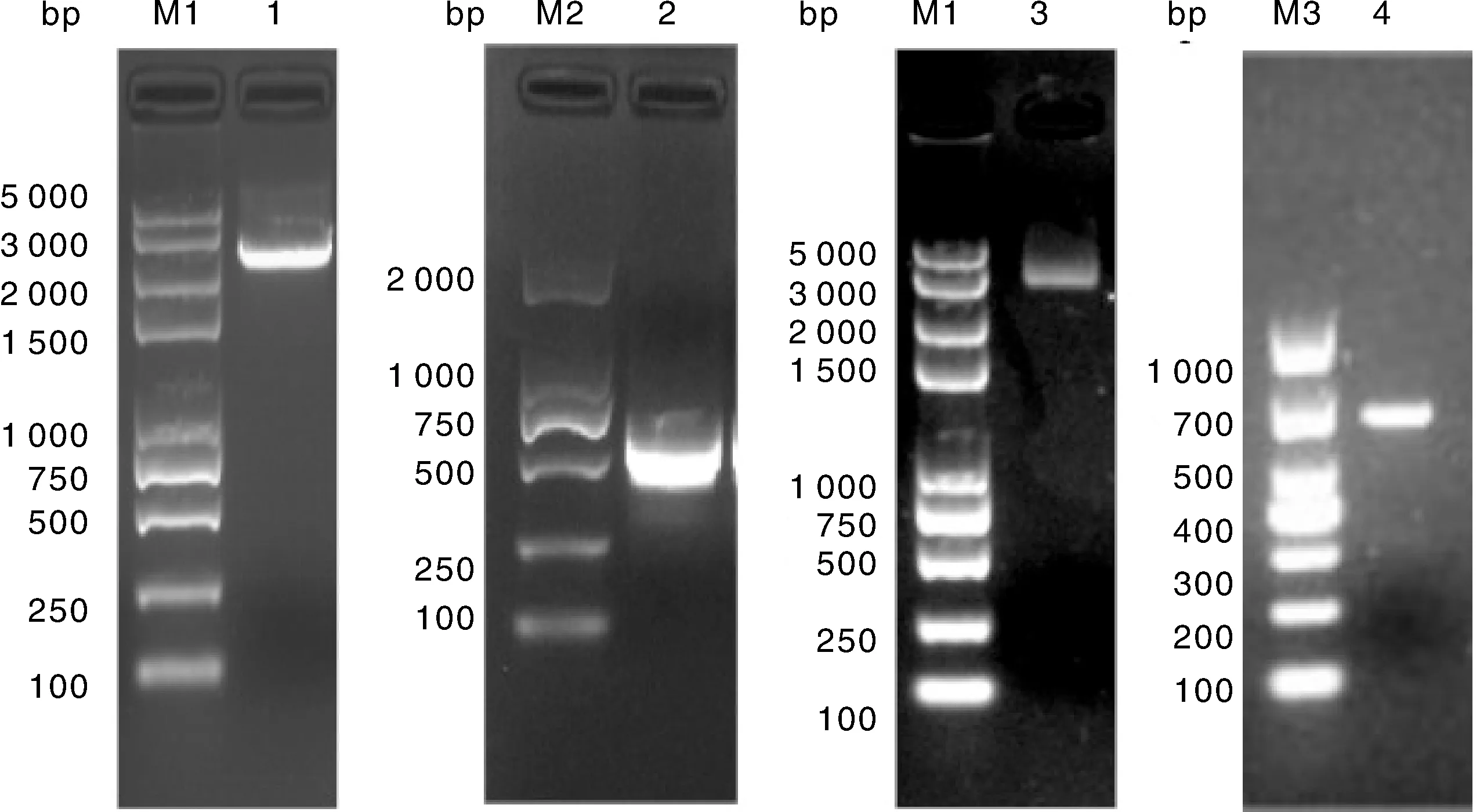
M1,DL5000 DNA分子量标准;1,pVAX1-1;M2,DL2000 DNA分子量标准;2,rCMV;3,pVAX1-2;M3,DL1000 DNA分子量标准;4,绿色荧光蛋白基因。M1, DL5000 DNA marker; 1, pVAX1-1; M2, DL2000 DNA marker; 2, rCMV; 3, pVAX1-2; M3, DL1000 DNA marker; 4, GFP gene.图3 prPgP载体目的片段PCR产物电泳图Fig.3 Electrophoresis of PCR products of PRPGP vector target fragment
2.2 重组质粒prPP和prPgP的酶切鉴定
将重组质粒prPP用SalⅠ、XmaⅠ双酶切,得到大小约2 966 bp和623 bp的两条带;将重组质粒prPgP用AflⅡ、NheI双酶切得到大小约3 582 bp和720 bp的两条带,均与预期结果一致(图4),说明载体构建成功。
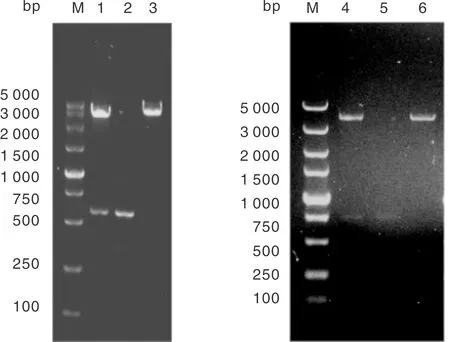
M,DL5000 DNA分子量标准;1,prPP用SalⅠ、XmaⅠ双酶切;2,rCMV;3,pVAX1-1;4,prPgP用AflⅡ、NheⅠ双酶切;5,GFP基因;6,prPP。M, DL5000 DNA marker; 1, Enzymatic cleavage of PrPP with SalⅠ and XmaⅠ; 2, rCMV; 3, pVAX1-1; 4, Enzymatic cleavage of prPgP with AflⅡ and NheⅠ; 5: GFP gene; 6: prPP.图4 prPP、prPgP重组质粒双酶切鉴定Fig.4 Identification of recombinant plasmid prPP and prPgP by double restriction enzyme digestion
2.3 同源臂gE L、gE R和E2的扩增结果
扩增的重组上下游同源臂gEL和gER产物大小和预期相符,分别为654、795 bp,BVDVE2基因大小约1 156 bp,与预期相符(图5)。
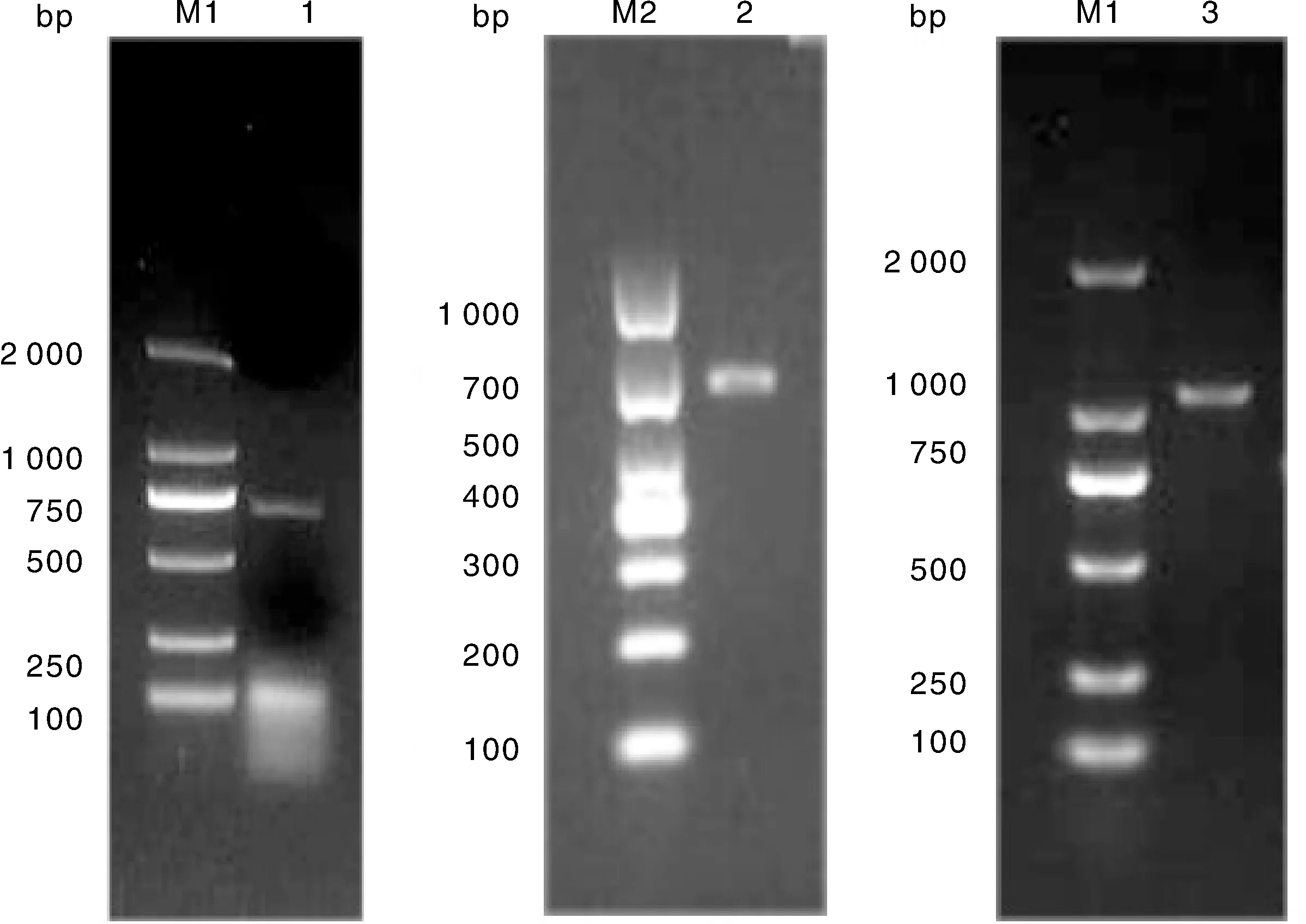
M1,DL2000 DNA分子量标准;1,gE L片段;M2,DL1000 DNA分子量标准;2,gE R;3,E2基因。M1, DL2000 DNA marker; 1, gE L; M2, DL1000 DNA marker; 2, gE R; 3, E2 gene.图5 p△gErPgP-E2质粒目的片段PCR产物电泳图Fig.5 Electrophoresis of PCR product of p△gErPgP-E2 plasmid target fragment
2.4 重组质粒p△gErPgP-E2的酶切鉴定
将p△gELrPgP用BamHⅠ、HindⅢ双酶切,得到大小约4 302 bp和654 bp的两条带;将p△gErPgP用PstⅠ、EcoRⅤ双酶切得到约4 956 bp和795 bp的两条带,将p△gErPgP-E2用BglⅡ、PstⅠ双酶切得到约5 751 bp和1 156 bp的两条带,所有酶切产物大小均符合预期(图6)。
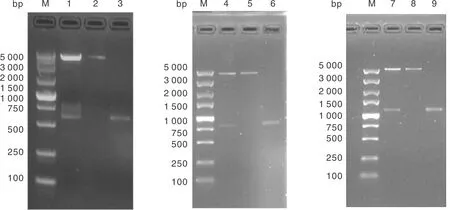
M,DL5000 DNA分子量标准;1,p△gELrPgP用BamHⅠ、HindⅢ双酶切;2,p△gELrPgP;3,rCMV;4,p△gErPgP用PstⅠ、EcoRⅤ双酶切;5,p△gErPgP;6,gE R片段;7,p△gErPgP-E2用BglⅡ、PstⅠ双酶切;8,p△gErPgP;9,BVDV E2。M, DL5000 DNA marker; 1, Enzymatic cleavage of p△gELrPgP with BamHⅠ and HindⅢ; 2, p△gELrPgP; 3, rCMV; 4, Enzymatic cleavage of p△gErPgP with PstⅠ and EcoRⅤ; 5, p△gELrPgP; 6, gE R; 7, Enzymatic cleavage of p△gErPgP-E2 with BglⅡ and PstⅠ; 8, p△gErPgP; 9, BVDV E2.图6 p△gErPgP-E2质粒双酶切鉴定Fig.6 Identification of p△gErPgP-E2 plasmid by double restriction enzyme digestion
2.5 重组载体的测序结果
重组质粒prPP、prPgP和p△gErPgP-E2经DNA测序,结果和预期一致。
2.6 重组病毒的纯化结果
重组病毒培养液经10倍系列稀释、连续传代筛选后,在荧光显微镜下不同代次重组病毒的绿色荧光差异明显。随着培养代次的递增,表达GFP的重组病毒占比逐渐增加,培养至第9代后得到全部呈现荧光的重组毒株rBHV-1-△gE/E2(图7)。
2.7 重组病毒的PCR鉴定
将培养至第9代的重组病毒rBHV-1-△gE/E2接种于MDBK细胞中进行增殖,提取病毒基因组,用引物扩增缺失的gE基因,没有获得目的条带;扩增GFP标记基因和外源抗原E2基因获得了预期的扩增条带(图8)。鉴定结果表明,重组病毒rBHV-1-△gE/E2中的gE基因已经缺失,并携带了标记基因GFP和外源抗原基因E2,同时证明重组病毒中不含亲本病毒,获得了rBHV-1-△gE/E2的纯培养物。
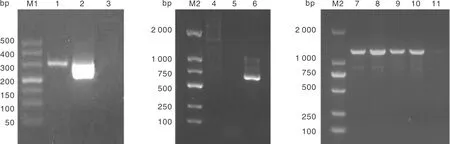
M1,DL500 DNA分子量标准;1,重组病毒GFP鉴定;2,GFP阳性对照;3,GFP阴性对照;M2,DL2000 DNA分子量标准;4,重组病毒缺失gE片段鉴定;5,gE阴性对照;6,gE阳性对照;7、8,重组病毒E2鉴定;9、10,E2阳性对照;11,E2阴性对照。M1, DL500 DNA marker; 1, Identification of GFP in recombinant virus; 2, GFP positive control; 3, GFP negative control; M2, DL2000 DNA marker; 4, Deletion identification of gE fragment in recombinant virus; 5, gE positive control; 6, gE negative control; 7 and 8, Identification of E2 in recombinantvirus; 9 and 10, E2 positive control; 11, E2 negative control.图8 重组病毒PCR鉴定Fig.8 PCR identification of recombinant virus
2.8 TCID50测定
根据测定结果(表4、表5),BHV-1亲本病毒滴度约为104TCID50·μL-1,重组病毒滴度约为103.5TCID50·μL-1。

表4 BHV-1的TCID50测定结果
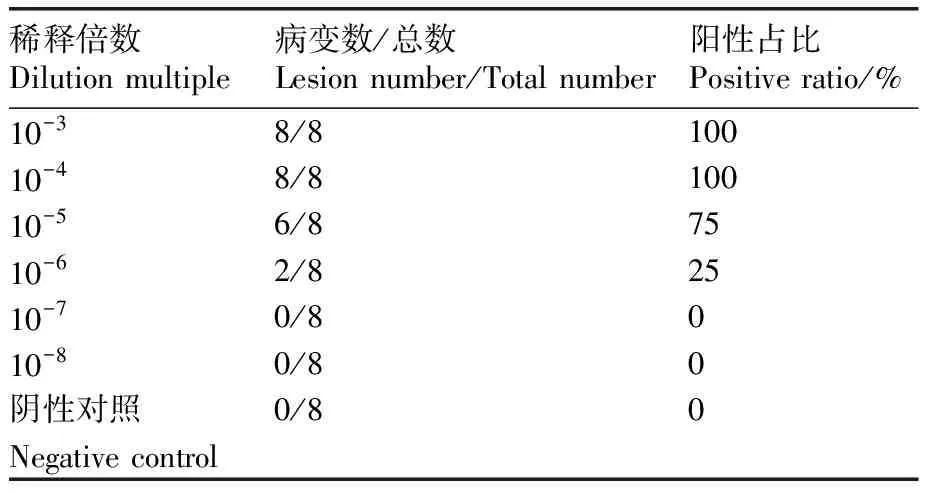
表5 rBHV-1-△gE/E2的TCID50测定结果
2.9 体外生长曲线测定
病毒生长曲线如图9所示,亲本病毒和重组病毒没有明显差异,证明gE基因缺失和外源基因的插入对病毒增殖无显著影响。
2.10 BVDV E2表达的鉴定
将重组病毒rBHV-1-△gE/E2接种MDBK细胞 24 h后,进行IFA试验。荧光显微镜下可见重组病毒感染细胞后呈现明亮的红色荧光,表明E2基因成功表达(图10)。
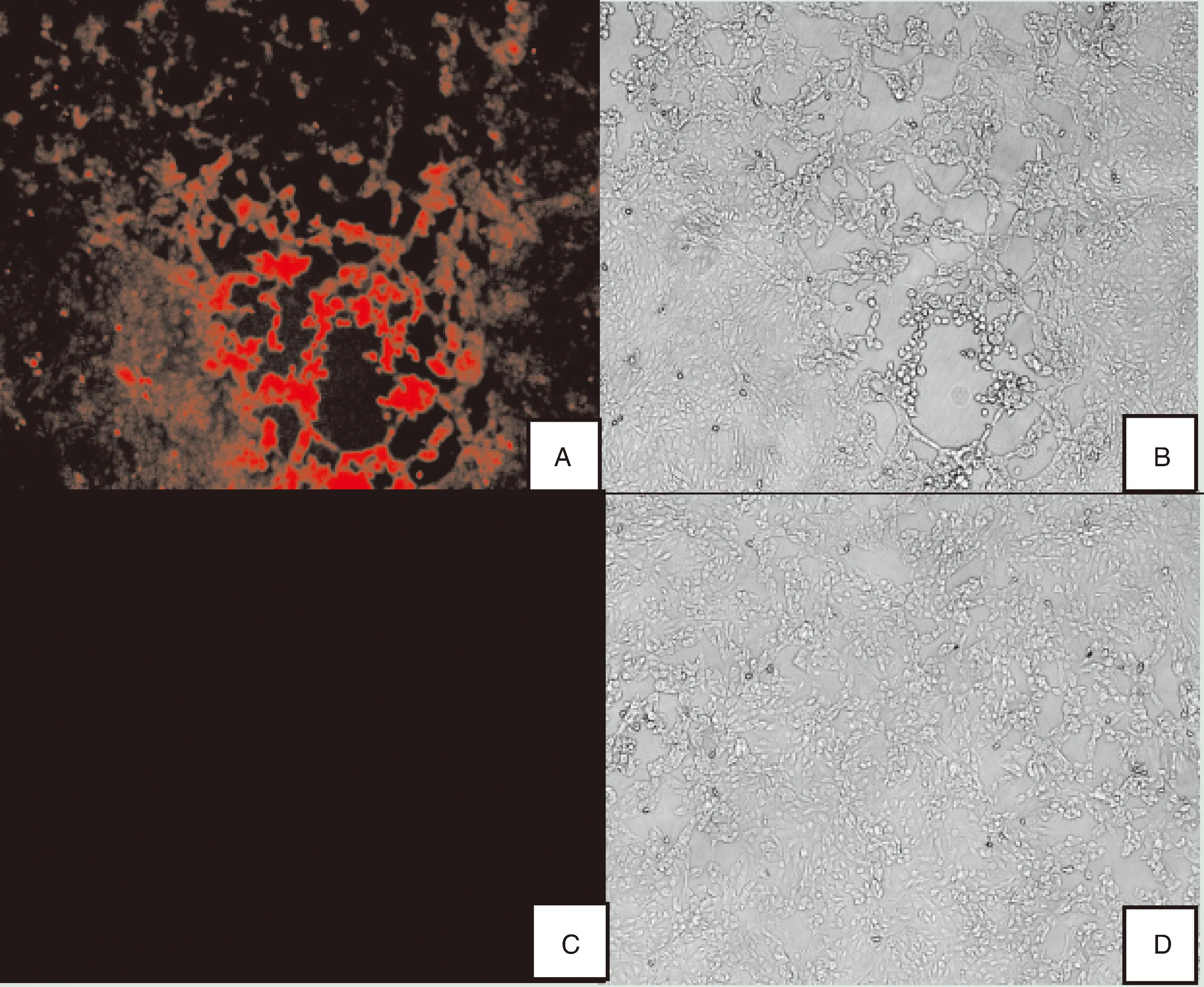
A,重组毒株rBHV-1-ΔgE/E2感染细胞的荧光情况;C,亲本病毒BHV-1感染细胞的荧光情况;B、D分别是A、C对应的明场下细胞形态。A is the fluorescence of recombinant strain rBHV-1-ΔgE/E2 infected cells; C is the fluorescence of parental virus BHV-1 infected cells; B and D are the cell morphology under bright field corresponding to A and C, respectively.图10 重组病毒接种MDBK细胞IFA结果(100×)Fig.10 IFA result of MDBK cell inoculated with recombinant virus (100×)
3 讨论
随着分子生物学技术的发展,基因工程疫苗技术日渐成熟,其中病毒活载体疫苗是当今与未来疫苗研制与开发的重要方向之一[4]。BHV-1作为活病毒载体具有多种优势,包括安全性好、能够插入多个外源基因、宿主范围窄、能够将病毒抗原直接呈递到黏膜表面激发特异性的黏膜免疫反应等[9]。所以,用BHV-1作为活病毒载体研制疫苗来控制牛病具有很大优势。
经同源重组产生重组病毒需要构建一个重组转移载体,这个载体必须包含与重组位点一致的上下游同源臂,根据需要还可携带外源基因[10]。鉴于以往BHV-1重组转移载体的使用范围不够广泛,每次重组都需要重新构建转移载体,笔者设计、构建了一个可广泛应用于重组BHV-1的转移载体。在质粒pVAX1 CMV启动子的反向位点引入一个反向CMV启动子,通过PCR反应在反向启动子下引入多克隆位点,构建了能够实现双向表达外源基因的载体prPP,进一步在反向启动子下插入GFP标记基因,方便重组病毒的筛选,所得转移载体命名为prPgP。由于在正、反向启动子下都有多克隆位点,prPgP可以方便地插入用于重组的同源臂序列,还可以根据需要选择不同的标记基因和外源抗原基因。
本研究利用构建好的转移载体prPgP,进一步构建在BHV-1gE基因位点进行同源重组的转移载体,以验证其应用的便捷性。首先在prPgP中插入重组位点的同源臂gEL和gER,利用同源重组将BHV-1中的gE基因缺失,同源臂的长度需控制在1 000 bp左右;长度过长会导致后续转染试验受到影响,过短则会影响BHV-1基因组识别[11]。构建好的转移载体p△gErPgP-E2携带了GFP标记基因和BVDVE2抗原基因,标记基因和目的基因分别位于gE缺失部位中2个方向相反的启动子下,独立控制2个外源基因的表达,互不干扰,保证了表达后重组蛋白的天然结构[12]。对于本研究构建的转移载体,今后可通过优化同源臂的长度,改变标记基因和外源基因的种类进行进一步的优化,扩大其应用范围。例如王银[13]利用BHV-1表达BVDV的E2基因,冯军科等[14]利用BHV-1表达牛呼吸道合胞体病毒G蛋白基因,任宪刚等[15]利用BHV-1表达O型口蹄疫病毒VP1基因等,这些都可以用prPgP转移载体实现。此外,本研究构建的转移载体还可以用于不同BHV-1基因缺失毒株的构建,为相关基因功能的研究提供方便。
4 结论
本研究构建的prPgP含有2个独立的CMV启动子和多克隆位点,可方便地用于BHV-1不同位点的同源重组,以研究BHV-1的基因功能;同时获得了能够独立表达2个外源基因的重组BHV-1,后续可用该病毒研制活载体疫苗,达到“一苗多用”的疫病防控目的。
