酪氨酸羟化酶基因新型突变所致多巴反应性肌张力障碍1例☆
2023-10-30储建敏吴蕾江璐璐王永利陈蔚欣陈玲
储建敏 吴蕾 江璐璐 王永利 陈蔚欣 陈玲
多巴反应性肌张力障碍(dopa-responsive dystonia,DRD)是由多巴胺合成通路遗传缺陷引起的疾病[1-3]。DRD的主要致病基因为编码三磷酸鸟苷环化水解酶(guanosine triphosphatecyclohydrolase,GTPCHI)的GCHI基因,呈常染色体显性或隐性遗传[4-6]。其次为编码酪氨酸羟化酶(tyrosine hydroxylase,TH)的TH基因和编码墨蝶呤还原酶(se⁃piapterin reductase,SPR)的SPR基因,均呈常染色体隐性遗传[7-8]。我国DRD患者中TH基因最常见的突变是p.Arg233His(突变频率为21.4%)和p.Gly247Ser(突变频率为21.4%)[9]。本文报告1例散发DRD病例,该病例为TH基因复合杂合变异所致,其中基因位点既往曾有报告,但其突变形式为新型突变。
1 临床资料
1.1 病例资料患者,女,29岁,体质量70 kg。因“肢体震颤伴手足姿势异常19年”于2019年1月6日就诊于我院。患者于2000年(10岁)紧张时出现四肢震颤,伴行走困难。心情平静后缓解。此后持物时亦有震颤,紧张时明显,早晨起床后症状较轻,入睡后消失。症状逐渐加重,可伴双侧手指屈曲呈爪样,活动手指可缓解;久行后出现双足跖屈、足尖着地,休息后改善。2009年患者出现行走时右侧拖步,遂于外院就诊,考虑“帕金森病”,予以“多巴丝肼62.5 mg每日1次,苯海索1 mg每日1次”药物治疗,症状基本缓解,此后维持该用药方案8年。2017年患者症状加重,震颤频率较前增多,右侧拖步较前明显,无伴手足姿势异常,于当地医院调整用药(具体方案不详)后好转。2019年症状再次加重,并至我院门诊就诊。病程中,患者无肢体麻木无力、肌肉萎缩、言语及吞咽困难等,无智能减退、嗅觉减退、幻觉、妄想、睡眠障碍等。既往史:发现结节性甲状腺肿数月,甲状腺功能正常。否认服用氟桂利嗪、吩噻嗪、利血平、曲美他嗪等药物。个人史无特殊。未婚未育,月经史无特殊。家族史:排行第一,有一弟弟,父母、弟弟体健(图1)。否认家族成员近亲婚配,否认存在相似症状家族成员。神经系统体检:卧立位血压正常,神清言利,颅神经查体无明显异常。肌张力不高;无肌肉萎缩,四肢肌力5级;步态正常。双侧快速轮替运动稍欠灵活,双侧指鼻试验、跟膝胫试验等共济运动完成好。双上肢轻微姿势性震颤,双上肢静止性震颤。感觉系统查体未见明显异常,双侧腱反射阳性(++),双侧病理征阴性。辅助检查:血常规、电解质、肝肾功能、铜蓝蛋白、甲状腺功能无明显异常。患者脑PET/CT显示:脑18氟-多巴(18F-DOPA)显像示双侧纹状体多巴胺代谢未见明显异常,提示双侧纹状体多巴胺神经功能正常;脑葡萄糖代谢未见明显异常。患者父母及弟弟未行脑PET/CT。
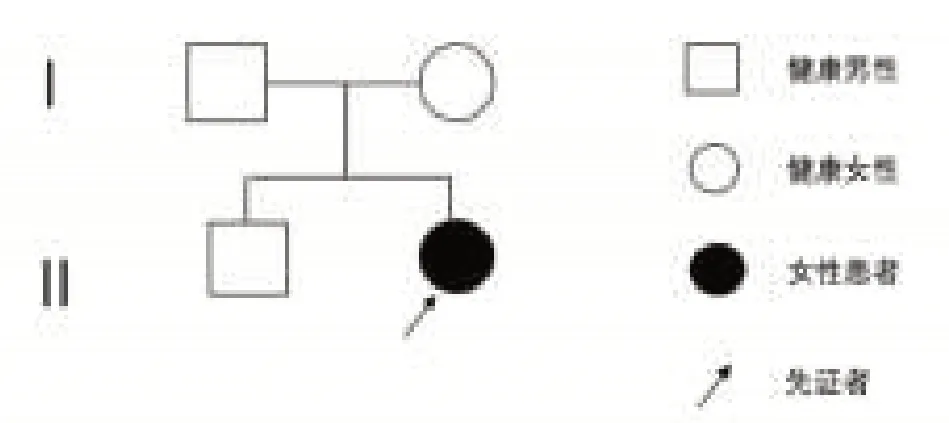
图1 患者家系
1.2 基因检测对患者及其父母进行高通量测序(患者弟弟未行基因检测)。鉴定出的致病基因突变位点,再通过Sanger测序在患者及其父母中进行验证。用乙二胺四乙酸(EDTA)抗凝管抽取患者及父母外周静脉血各2 mL,采用QIAamp DNA提取试剂盒(QIAGEN公司)抽提基因组DNA,并测量其吸光度值及浓度。提取的DNA用DNA酶片段化后用磁珠法进行纯化,随后进行PCR扩增并连接上接头序列,经TruSight One Sequencing Panel(illumina Inc,USA)两次捕获及纯化,再经PCR扩增和纯化后获得最终文库,在NextSeq500测序仪(illumina Inc,USA)对40个临床相关基因(采用广州金域医学检验中心设计的肌张力障碍相关基因包)的外显子区进行测序。基因检测结果提示,患者存在TH基因EXON9 C.943G>A错义突变及TH基因EXON8 C.851A>G错义突变的杂合突变,患者父亲TH基因EXON9 C.943G>A错义突变,患者母亲TH基因EXON8 C.851A>G错义突变(图2、3)。

图2 患者TH基因检测结果

图3 患者父亲及母亲TH基因检测结果
1.3 诊断与治疗通过患者的临床表现、查体以及基因检测结果,最终明确诊断为多巴反应性肌张力障碍。治疗上予以多巴丝肼62.5 mg每日1次(早餐饭前1 h服用),并建议患者若夜间运动症状加重,可在晚餐前加用多巴丝肼62.5 mg。后续对患者进行了3年随访,末次随访时间为2022年2月24日。随访过程中,患者病情可控,震颤较前减少,服药后未出现异动、开关现象及剂末现象等。
2 讨论
本例患者存在肢体震颤及手足姿势异常,且症状具有昼夜波动性,经小剂量左旋多巴治疗有效且疗效持续,符合DRD的临床特点。基因检测结果显示,患者存在TH基因EXON9 C.943G>A错义突变及TH基因EXON8 C.851A>G错义突变。TH基因位于Chr11p15.5,编码的TH为多巴胺合成限速酶[7,10]。TH基因突变导致其酶活性降低,直接影响多巴胺及其下游产物的合成,从而引起一系列儿茶酚胺类物质缺少的临床症状[11],如肌张力障碍、运动功能减退、动作迟缓、肢体僵硬、震颤、动眼危象、上睑下垂等[12-15]。部分患者还可出现抑郁、睡眠质量降低等非运动症状[16]。2022年WEISSBACH等[17]研究发现,TH基因相关DRD多见于亚洲人群(57%)及欧洲白人(19%),且其中有32%的TH基因相关DRD患者有阳性家族史。左旋多巴对94%的TH基因相关DRD治疗有效(平均治疗剂量为208 mg/d或5.0 mg/kg/d)。WIJEMANNE等[18]将TH基因相关性DRD分为A型及B型两种类型:A型患者主要表现为锥体外系运动症状,对左旋多巴反应良好;B型临床症状较为严重,起病年龄更早,以明显的运动发育迟缓、喂养困难、肌张力异常为主要表现,且对左旋多巴反应较差,几乎均伴随智能、运动发育问题。结合本病例特点,患者可归类为A型。根据2021年的一项研究,TH基因突变引起的DRD患者需要个体化治疗方案[19],本例患者左旋多巴治疗剂量为50 mg/d,即0.71 mg/kg/d,患者症状控制良好。
本病例中的C.943G>A位点突变,首次在酪氨酸强化酶缺乏症患者中报告[20]。随后我国一项家系研究表明,该基因突变可导致家族性DRD[21]。此后的研究发现,存在该基因变异的DRD患者多表现为婴幼儿起病,可有四肢无力、智能发育迟缓、肌张力障碍、对小剂量左旋多巴反应较好等临床特点[22-23]。另一突变位点C.851A>G会导致相关基因编码的蛋白第284位氨基酸由谷氨酸转为甘氨酸,但目前与之相关的临床病例尚未见文献报告,故该基因突变是否具有致病性有待进一步研究。
既往欧洲及我国文献报告TH基因相关DRD的最热门突变位点为C.698G>A,C.457C>T突变频率为第2位[7,22,24]。其中C.457C>T可能为中国人TH基因的热点突变,并且该位点与不良预后有关,如起病年龄小于1岁,且有该位点突变,临床预后可能不佳,具体原因有待进一步研究[22]。本例患者的突变位点为C.943G>A及C.851A>G,既往文献尚未发现C.943G>A位点突变与临床预后的关系,C.851A>G位点突变目前尚无报告。在随访中,本患者症状控制尚平稳,有轻微运动迟缓,不影响生活。
综上所述,DRD是一种遗传性肌张力障碍疾病,临床上表现为肌张力障碍的疾病多种多样,遗传学检测有利于早期明确诊断。TH基因变异所致DRD临床表型分为A型与B型两类,需通过患者的临床表现、起病形式及长期随访来确定。对于TH基因突变的患者来说,A型患者对左旋多巴有更好的反应。
