金属氧化物与掺氮生物质协同吸附CO2机理
2023-10-20汪辉春顾明言
汪辉春,王 莹,顾明言,陈 萍
(安徽工业大学 能源与环境学院,安徽 马鞍山 243002)
0 引 言
全球变暖日益严重,温室气体排放量增加与全球变暖、生态恶化直接相关。作为温室气体中含量较多,占比最大的CO2,其获取、储存和利用对于改善全球气候、实现其价值具有重要意义。如何有效捕获和存储CO2是关键。多孔固体吸附具有低成本、低能源要求、操作简单、工艺清洁等优点,是一种很有前途的替代方法,该技术是具有优异CO2吸附特性的固体材料。目前对众多捕获CO2的多孔固体吸附剂研究较多,如多孔碳[1]、沸石[2]、金属有机框架(MOFs)[3]和多孔聚合物[4]等。沸石可在几分钟内达到其吸附能力,然而气体中存在H2O时,其吸附能力大幅降低。当CO2含有H2O被沸石吸附时,酸性条件可能导致沸石结构脱铝,从而导致骨架部分或全部破坏,最终降低CO2吸附容量,而MOFs的稳定性和合成成本较高一直是其实际应用的最大限制,多孔聚合物的合成通常需要复杂的单体、昂贵的催化剂或严格的条件,使合成过程昂贵、繁琐,规模扩大困难。近年来,多孔碳具有成本低、表面积大、孔隙率高,可调孔隙结构和表面特性,低能量下再生和高稳定性等特点[5-6],备受关注。用于合成多孔碳的各种前驱体中,生物质材料因其广泛的使用性、低成本、可再生和净零排放受广泛关注。
作为多孔碳吸附剂,提高生物质炭吸附特性是亟需开展的工作。研究表明表面积、孔体积、孔径、氮含量、氢含量等特性中,氮含量对多孔碳捕获CO2至关重要。研究表明,引入氮官能团可有效提高表面特性,从而提高对CO2吸附能力[7-10]。通过氢键[11]、酸碱[12]和四极-四极相互作用[13],增强吸附剂对CO2的吸附容量和选择性。YUE等[14]制备的氮掺杂碳在25 ℃时CO2吸附容量达3.71 mmol/g,与未掺杂样品相比增加40.53%;SETHIA等[15]合成的富氮活性炭在25 ℃下具有高CO2吸附容量(5.39 mmol/g)和高CO2/N2选择性。对于相同微孔结构的碳,N含量在CO2吸附中具有重要作用。
在掺氮多孔碳材料中,氮官能团以吡啶、胺/亚胺、吡咯、季氮和氧化氮形式存在,进一步研究发现不同氮基团对CO2吸附贡献不同。XU等[16]在650 ℃ 下,通过尿素掺杂葡萄糖,在KOH活化作用下制取多孔碳,得到的吡咯氮高于吡啶氮和季氮,推测吡咯氮可能有利于CO2捕获。SNCHEZ-SNCHEZ等[17]研究了不同氮在CO2吸附中的作用,发现N-6、N-5和G-N类型的氮基团有利于CO2吸附,但N-5对CO2捕获的贡献高于N-6和G-N。WANG等[18]用密度泛函理论(DFT)计算描述了CO2与多孔碳吸附的相互作用,发现含吡咯氮碳表面与CO2存在分子-分子间色散作用,增强了氮掺杂碳基表面对CO2的吸附性能。综上所述,不同氮基团对CO2吸附特性的影响尚未明晰。
多孔碳材料仍面临碳表面缺乏高活性吸附位点的缺陷。研究表明,多孔碳材料的吸附能力与其丰富的分层孔隙结构密切相关[19-21]。表面活性基团改性是提高CO2吸附能力的有效方法[22-24]。研究表明金属可促进生物炭孔隙生成,增强表面极性。陈伟[25]在富氮热解过程中引入KOH为活化剂,高温下(800 ℃),NH3和KOH均具有活化作用,相互促进并剧烈刻蚀碳骨架,促使生物质炭生成大量孔隙,并形成丰富的活性空位富集大量氮,从而形成具有高比表面积、发达介孔的掺氮碳材料。但目前对于不同金属与掺氮炭材料协同作用下对CO2吸附效果影响的研究鲜有报道。
基于此,笔者通过密度泛函理论计算研究掺氮生物炭、金属氧化物和掺氮生物质炭协同金属氧化物的吸附性能。研究了掺氮生物炭中不同类型的含氮官能团、金属氧化物及二者协同作用下对CO2吸附效果的影响,进而为生物质炭掺氮基团和金属耦合掺氮生物质炭的定向调控制备提供支撑。
1 密度泛函理论计算
密度泛函理论(Density Functional Theory,DFT)是一种研究多电子体系电子结构的量子力学方法。密度泛函理论在物理和化学上应用广泛,用来研究分子和凝聚态的性质,是凝聚态物理计算材料学和计算化学领域最常用的方法之一。
通过密度泛函理论[26]计算研究不同掺氮生物炭(吡啶氮(N-6)、吡咯氮(N-5)、芳香氮(G-N)、吡啶氮氧化物(N-X))、金属氧化物(CaO、MgO)及金属掺杂富氮生物炭对CO2的吸附效果(考虑到实际吸附时,生物质炭与金属混合制样后吸附在同一环境温度下进行,模拟计算活性炭、CaO/MgO单独吸附CO2及协同作用下不考虑温度)。所有DFT计算均使用Materials Studio软件CASTEP模块[27]。GGA(广义梯度近似)方案和PBE(Perdew-Burke-Ernzerhof)泛函用于描述交换和相关相互作用。利用超软赝势描述电子和离子核间相互作用。选择自旋极化选项进行自旋不受限制计算。几何优化选择BFGS优化算法。CaO和MgO均为立方晶体体系,为保证可比性,保持几何优化参数设置一致。能量在一个单体网格中k点收敛于6×6×6。电子波函数扩展为平面波,截止能量为380 eV。几何优化收敛准则为:自洽场5.0×10-7eV/atom,能量5×10-6eV/atom,位移5×10-5nm,力0.001 eV/nm,应力0.02 GPa。
1.1 掺氮生物质炭
研究发现高CO2吸附容量与多孔炭表面掺氮有关[28]。氮在生物质炭中的存在形态主要有吡啶氮(N-6)、吡咯氮(N-5)、芳香氮(G-N)、吡啶氮氧化物(N-X)[29-31],为探究不同含氮基团对生物炭表面NO吸附能力的影响,分别构建吡啶氮生物炭(CN-6)、吡咯氮生物炭(CN-5)、芳香氮生物炭(CG-N)及吡啶氮氧化物生物炭(CN-X),具体模型结构[10]如图1所示。N-6与N-5的吸附位点主要分布在炭层边缘;G-N有4个外层电子,其中4个电子可形成σ键和π键,近邻碳原子位点;N-X通常存在于碳材料骨架内。故在生物质炭表面引入了4种含氮官能团,在4种掺氮生物质炭表面进行吸附计算,比较不同吸附方式对CO2吸附的影响。
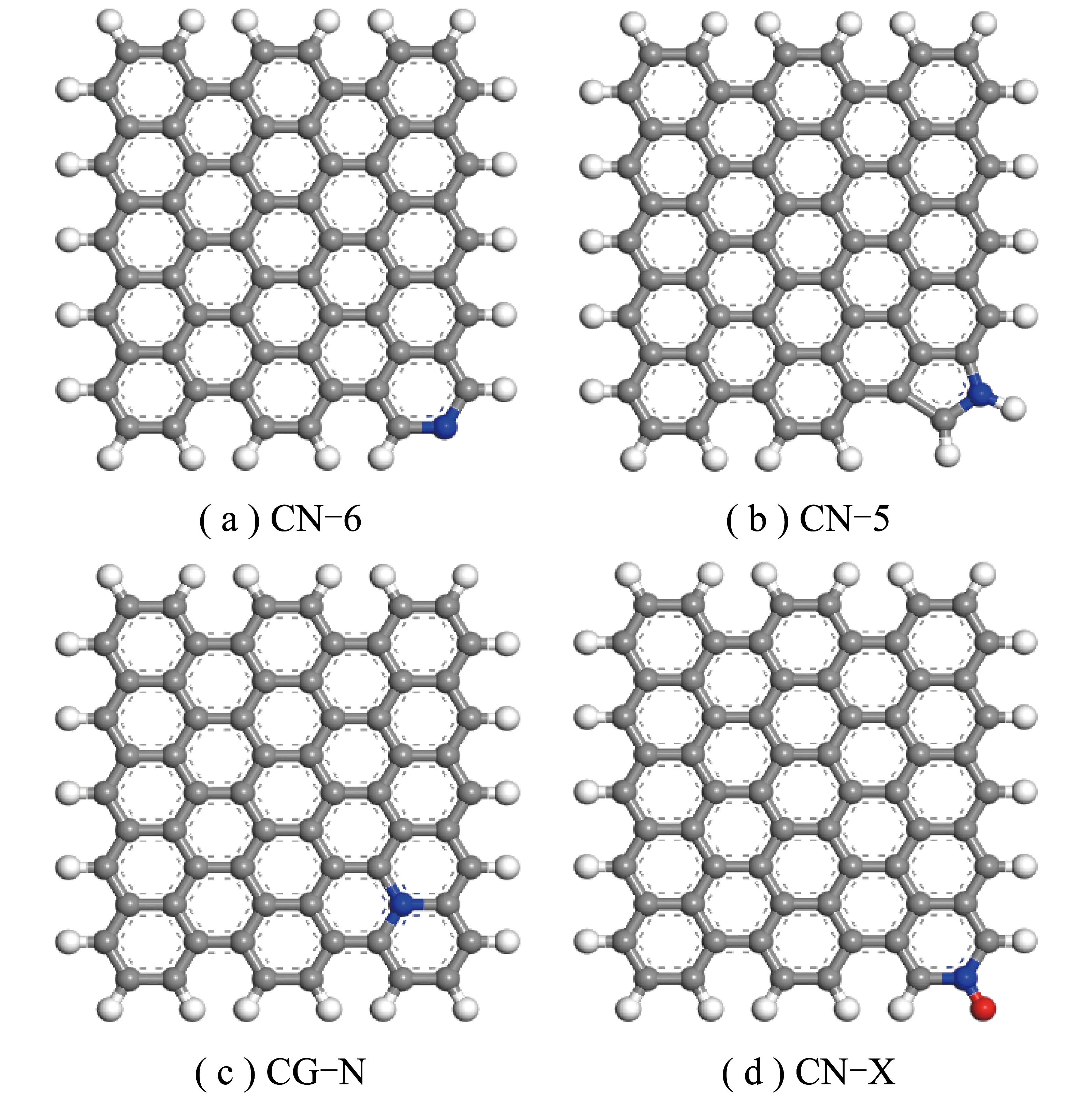
图1 掺氮生物质炭模型结构Fig.1 Model structure of nitrogen-doped biochar
1.2 金属氧化物
CaO与MgO试验及模拟的原胞晶格参数[29-30]见表1,其中CaO参数相对误差分别为0.09%和0.51%,几何优化后的氧化钙晶格参数与之前试验结果一致。证明优化CaO、MgO具有准确性。

表1 原胞晶格参数Table 1 Protocyte lattice parameters
CaO与MgO均为立方晶体、空间群一致,以CaO为例(图2,长、宽、高分别为1.024 93、1.024 93、2.466 31 nm)。CaO属于Fm-3m(225)空间群数和岩盐结构的立方晶体体系。CaO(001)是一个典型的表面,可有效描述CaO与气体CO2间的固、气反应,对CaO原胞001面进行切面,切面厚度设置为5层。真空区域设置为1.5 nm,以避免周期性图像间的相互作用[31]。切面表层松弛,固定其余4层原子。由图2可知,通过a层b×b表面板模型建模描述CaO(a=5,b=3)。平板模型和相关吸附结构的能量在Monhorst-pack网格中k点收敛于3×3×1。
图2 CaO模型Fig.2 CaO model
吸附能Eads为
Eads=Epro-(Eslab+Eadsorbate)。
(1)
其中,Esds为吸附质在吸附剂表面吸附能;Epro为吸附产物总能量;Eslab为平板模型总能量;Eadsorbate为分离的吸附质CO2在其平衡几何形状下的总能量。Eads为负,对应放热过程,该值越小,释放的热量越多,吸附越强,表示一个稳定的吸附系统。通过计算CO2吸附在吸附剂表面的吸附能可比较吸附效果。
2 结果与讨论
2.1 生物质炭各吸附位点
CO2在生物质炭表面的吸附方式主要有平行吸附和垂直吸附,为更好比较含氮官能团对CO2吸附效果,采用与生物质炭表面同样的吸附方式在掺氮生物质炭表面进行CO2吸附。4种含氮官能团生物质炭对CO2的不同吸附方式及吸附高度如图3所示,吸附相关原子笛卡尔坐标见表2,为便于介绍,对相关原子进行编号,以N-X为例(图4):将吸附的CO2中C命名为C66,O分别命名为O1、O2,N-X中氮原子命名为N,氧原子命名为O3,各吸附方式下的吸附能见表3。通过比较发现,与生物质炭相比,掺氮生物炭可提高CO2分子的吸附能力。
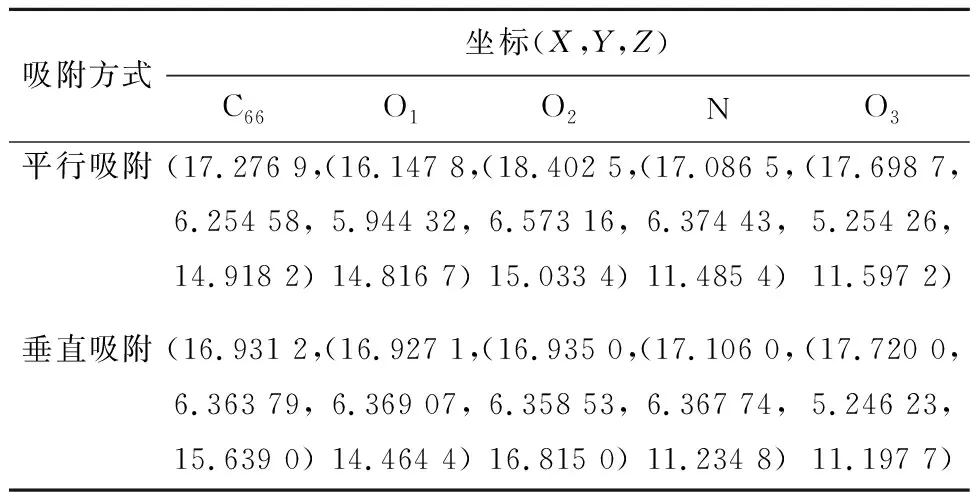
表2 掺N-X生物质炭对CO2吸附后的相关原子坐标Table 2 Related atomic cartesion coordinates of CO2 adsorption by N-X biochar

图4 相关原子编号Fig.4 Relevant atomic number
由于吡啶氮氧化物对CO2平行吸附的吸附能最低,将其确定为最稳定的CO2吸附构型。
2.2 生物质炭对CO2的吸附
为进一步说明4种含氮官能团对CO2平行吸附的吸附行为,对4种含氮官能团进行电荷差分和电荷分布计算。电荷差分计算结果如图5所示,电荷转移计算结果见表4~7,s、p及Total分别为各原子最外层s、p轨道上及最外层总电荷。

表4 N-6对CO2吸附过程Mulliken电荷分布情况Table 4 Mulliken charge distribution during the adsorption of CO2 by N-6
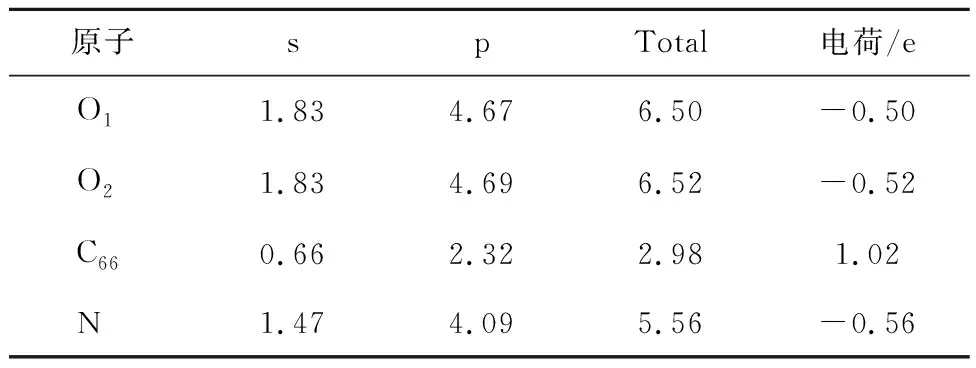
表5 N-5对CO2吸附过程Mulliken电荷分布情况Table 5 Mulliken charge distribution during the adsorption of CO2 by N-5
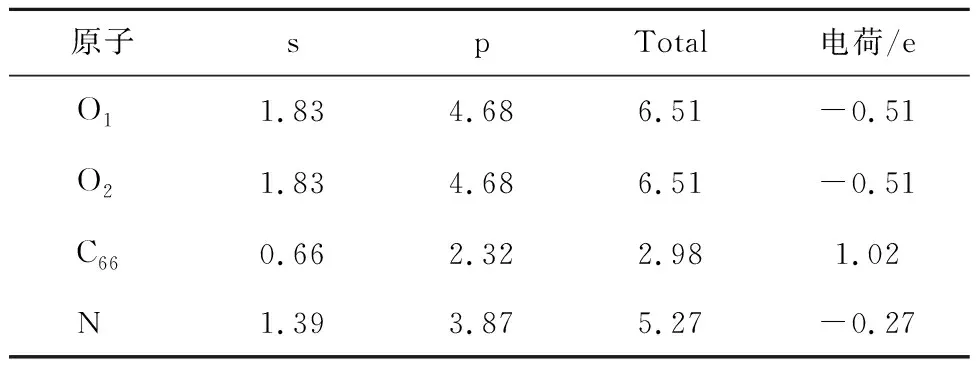
表6 G-N对CO2吸附过程Mulliken电荷分布情况Table 6 Mulliken charge distribution during the adsorption of CO2 by G-N
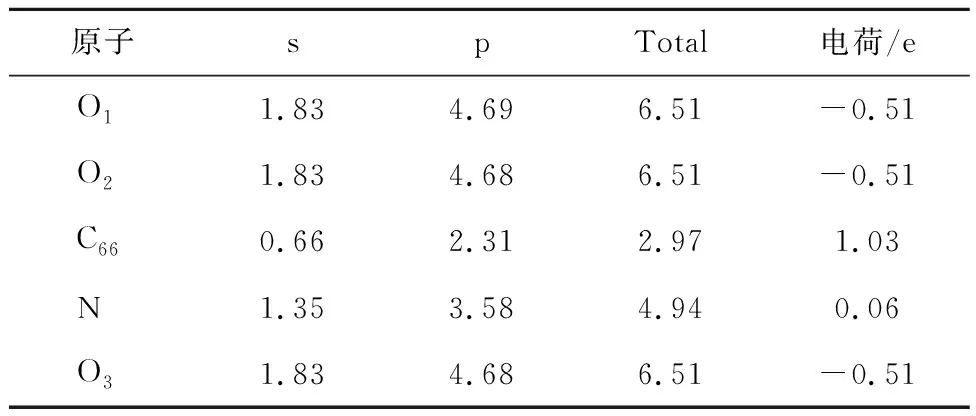
表7 N-X对CO2吸附过程Mulliken电荷分布情况Table 7 Mulliken charge distribution during the adsorption of CO2 by N-X
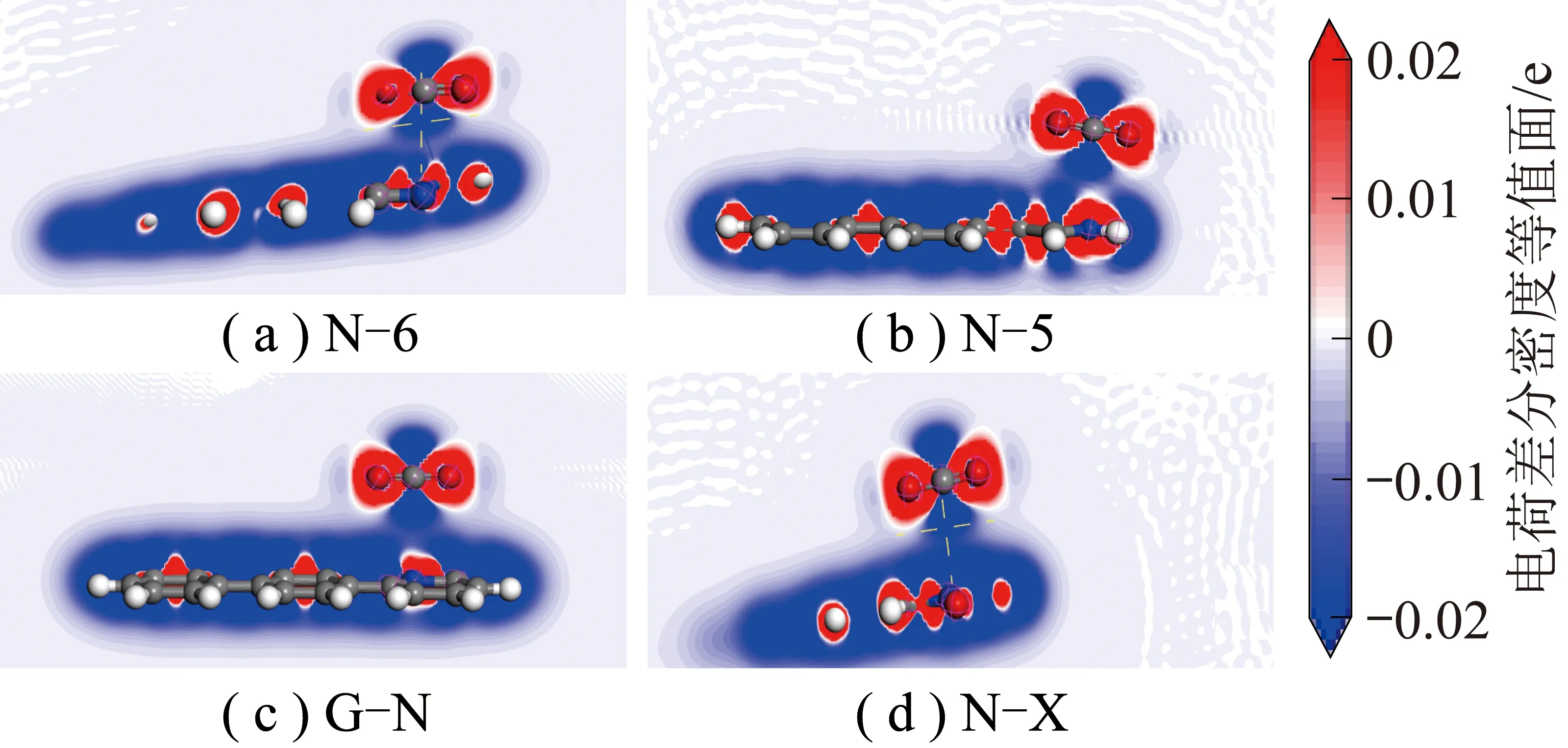
图5 N-6、N-5、G-N和N-X对CO2平行吸附的电荷差分密度Fig.5 Charge differential density with N-6,N-5,G-N and N-X adsorb CO2 in parallel
图5中红色、蓝色区域分别为得到和失去电子,由图5可知,N-5周围得到较多电子,其次是N-6,G-N周围得到较少电子,而N-X周围失去电子,表4~7得出相同结论。由表4~7可知,CO2分子对不同氮基团中N的影响不同。N-6、N-5、G-N中N与CO2中O呈同种电荷,N-X中N与CO2中O呈异种电荷,表明N-X与CO2间存在较其他氮基团更强的引力作用。基于N-X基团中N与O的电荷分布情况发现,CO2中O对N-X基团中N的作用力更强。表明N-X对CO2吸附影响较大,与CO2分子间存在弱相互作用力。因此采用含N-X生物质炭作为后续与金属氧化物耦合提供掺氮生物质炭模型。
2.3 金属氧化物各吸附位点吸附能计算
CO2在CaO表面发生非均相反应。分析CO2中C在MgO、CaO的001表面O位点3种吸附结构(分别为Top、Hollow、Bridge),如图6所示。
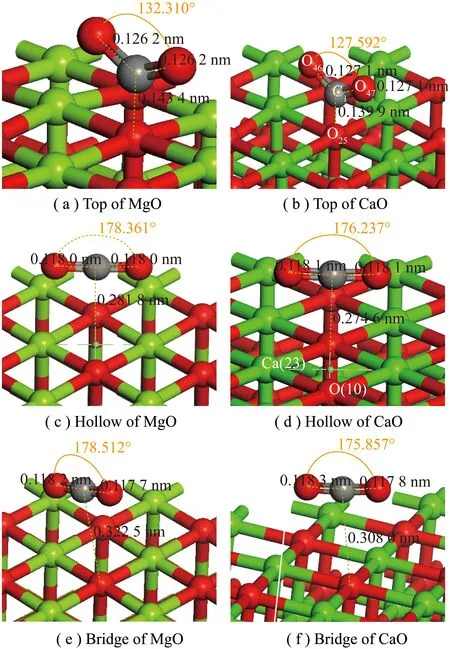
图6 CO2在MgO(001)和CaO(001)表面的Top、Hollow、Bridge吸附Fig.6 Adsorbed of CO2 on the surface of MgO(001) and CaO(001) Top,Hollow and Bridge
根据建模时软件对原子的编号(通过Label设置可视),用原子+下角标命名。C1:CO2中碳原子;O46、O47:CO2中氧原子;O25:CaO、MgO中O-Top吸附的原子。MgO、CaO吸附CO2前后原子坐标见表8、9。

表8 金属氧化物优化后相关原子坐标Table 8 Metal oxide optimized correlation atomic coordinates

表9 金属氧化物吸附CO2后相关原子坐标Table 9 Relevant atomic cartesion coordinates of metal oxides after adsorption of CO2
3种吸附结构收敛后的吸附能计算结果见表10。可知3种吸附结构中,O原子顶部吸附(O-Top)吸附能最大,吸附效果最好;进一步比较相同吸附结构,发现CO2在CaO的001表面上O-Top吸附结构的吸附能较MgO表面高-92.225 kJ/mol。该吸附结构下CaO表面吸附能为-136.697 kJ/mol,表明发生了化学吸附(吸附能小于-100 kJ/mol[32])。吸附能结果表明CO2在CaO表面O-Top吸附较稳定。
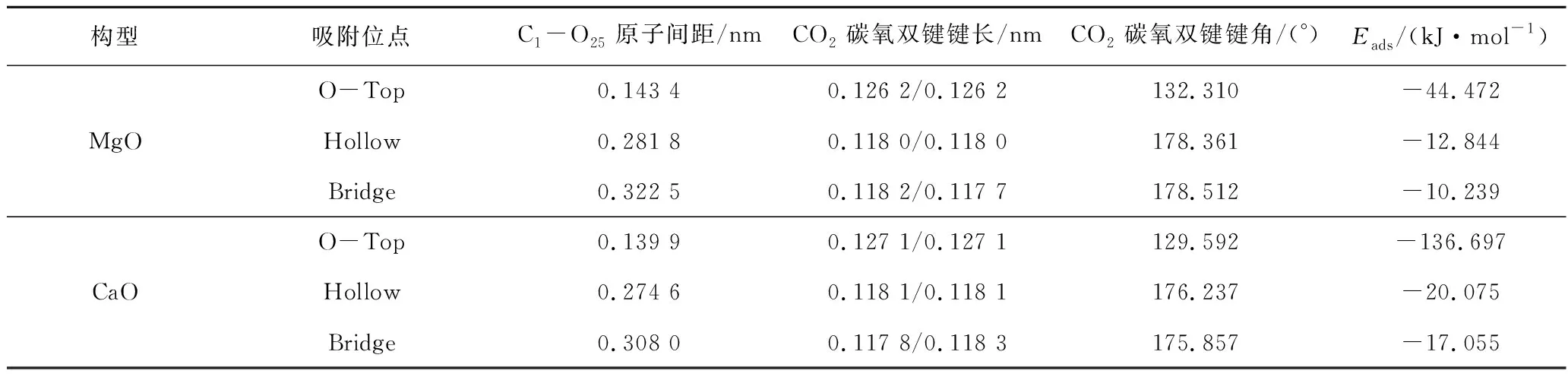
表10 各吸附位点吸附能Table 10 Adsorption energy at each adsorption site

比较CaO和MgO相同吸附位点的C1—O25键长、CO2中键长、键角及吸附能,发现C1—O25键长、CO2中键角与吸附能呈负相关,CO2中C1—O46、C1—O47键长与吸附能呈正相关。这可能是由于CaO较大的吸附能使CO2吸附后C1—O25键能更大,原子间作用引力更强;同时,C1—O46、C1—O47间作用力减弱。键角较小可能由于O46、O47之间斥力更小或Ca与O间斥力比Mg与O间斥力大。结合同一金属氧化物不同吸附方式仅O-Top的键角小,进一步推测是Ca、Mg与O间斥力引起键角变化。
2.4 金属氧化物对CO2吸附
为进一步阐明CO2分子在CaO(001)表面的电子相互作用、结合强度和电荷转移,研究了主要相互作用原子在其化学吸附位点上的分波态密度(Partial Density of States,PDOS)、电荷差分密度(Electron Density Difference,EDD)及布居分析(Band Populatin Analysis,BPA)。分波态密度可解释吸附质与吸附剂表面之间电子相互作用。分波态密度可探究吸附剂表面不同吸附构型对气体吸附情况及内在机理和电子性质。
CO2在CaO和MgO表面不同吸附构型分波态密度如图7所示。由图7(b)、7(c)、7(e)、7(f)可知,CO2吸附在CaO(001)和MgO(001)表面后,被吸附原子与表面位点之间无轨道杂化,说明在Hollow、Bridge吸附方式下,CO2与CaO(001)及MgO(001)表面的相互作用力较弱。图7(a)显示CO2分子C端吸附在CaO(001)表面的O位点后,吸附后的C1与表面的O25位点有明显杂化,在2s轨道的-22.40、-8.15 和-6.20 eV、2p轨道的-8.15、-6.20及4.5~11.2 eV大量重叠,反映了被吸附的C1和吸附表面O25间形成共价键。分波态密度的结果分析与吸附能分析结果一致,表现为O-Top吸附方式吸附效果最好。

图7 CaO和MgO表面Top、Hollow、Bridge吸附PDOS图Fig.7 Top,hollow and bridge adsorption PDOS on the surface of CaO and MgO
PDOS分析了C1与O25间的轨道杂化,得到的EDD图可直观分析电子的转移方向和分布强度。
CaO和MgO表面O位点上吸附的电荷差分密度如图8所示,Ca和Mg外部黄色区域均存在电子偏移(失去电子);相较Mg,Ca周围内部蓝色区域明显存在电子聚集(得到电子),而Mg周围未发现。进一步对电荷差分密度进行剖面分析,如图9所示(切割平面通过C1、O46、O47、O5,红色和蓝色区域分别代表得到和失去电荷)。结果表明被吸附CO2中电子由C1移向O46、O47,同时还移向O25;整体上CO2从CaO表面上得到电子,而O25从C1上得电子,推测吸附表面电子会由Ca移向O46、O47。C1和O25间重叠的电子云表明其之间发生很强的相互作用,且这2个原子成键。这种相互作用导致电子分布重排。
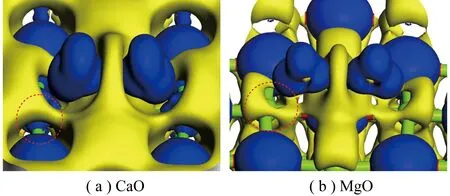
图8 CaO和MgO上O-Top吸附电荷差分密度Fig.8 Differential density maps of O-Top adsorption charge on CaO and MgO
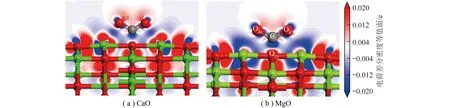
图9 CO2在CaO和MgO上吸附电荷差分密度截面Fig.9 CO2 adsorbs charge differential density cross-sections on CaO and MgO
从电子聚集区域比较CaO和MgO对CO2吸附影响,可知Ca中一部分电子向CO2偏移,另一部分电子被左侧和下侧氧原子吸引(蓝色区域),CaO吸附的CO2中O46、O47周围聚集电子较MgO更多。Ca与C1之间可能存在弱相互引力,使CO2更易被CaO吸附;而Mg内部电子收拢到原子核,外部电子偏移向四周的氧原子,形成Mg外部周围电子缺失情况。虽然EDD可定性展示电子转移方向和强度。但为从数值上阐述键强度和吸附稳定性,对CaO和MgO表面吸附CO2进行布居分析。布居分析可判断键强度,反映了原子周围电子布居的分配。布居值越大,共价键越强,相反,则离子键越强。
电荷转移和键布居计算结果见表11,对于CaO和MgO吸附CO2的O-Top构型,电子从CaO和MgO表面分别转移了-0.69和-0.59 e到CO2分子中;C1—O25键的键布居数分别为0.62和0.56 e。CaO中O25与C1间布居值大于MgO,则共价键更强,稳定性更好,较高的电子转移和键居数进一步验证了CO2的C与CaO表面O之间相互作用更强。这与电荷差分密度和吸附能分析结果相符。进一步证明CO2在CaO表面形成共价键。
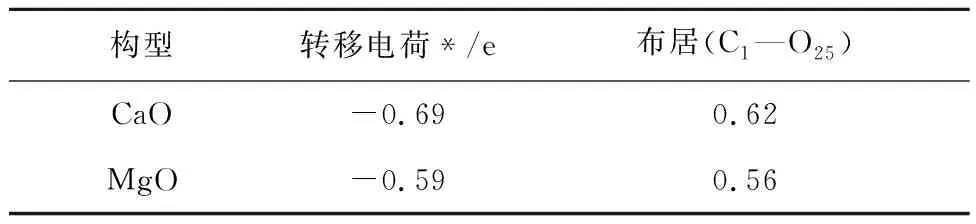
表11 布居分析Table 11 Population analysis
MgO与CaO对CO2吸附过程Mulliken电荷分布情况见表12、13,发现Mg、Ca、C1失去电子,O得到电子,得失电子求和为CO2得到0.63 e。Ca的M+N层、Mg的L+M层轨道电子之和(可失去最多电子)均为10。Ca比Mg多失去电子0.06 e,极性更强,对O46、O47吸附更强。相较Mg,Ca由于电子跃迁产生了新能级(存在d轨道),电子云区域更广,与O46、O47重叠更密。从电子转移数量角度印证EDD分析得出的电子转移结论,说明CaO对CO2的吸附优于MgO。
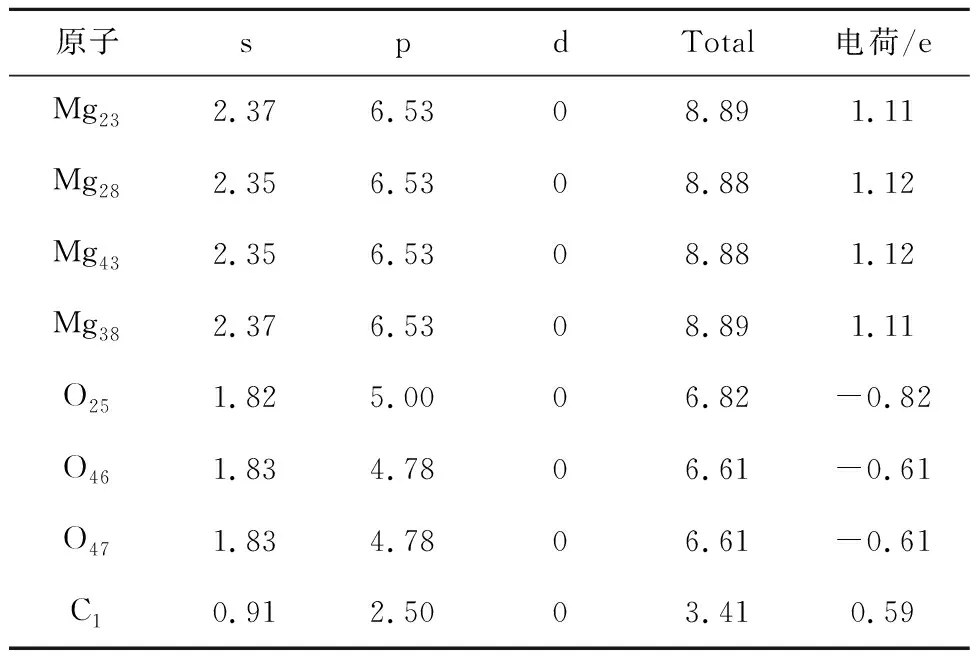
表12 MgO对CO2吸附过程Mulliken电荷分布情况Table 12 Mulliken charge distribution during the adsorption of CO2 by MgO
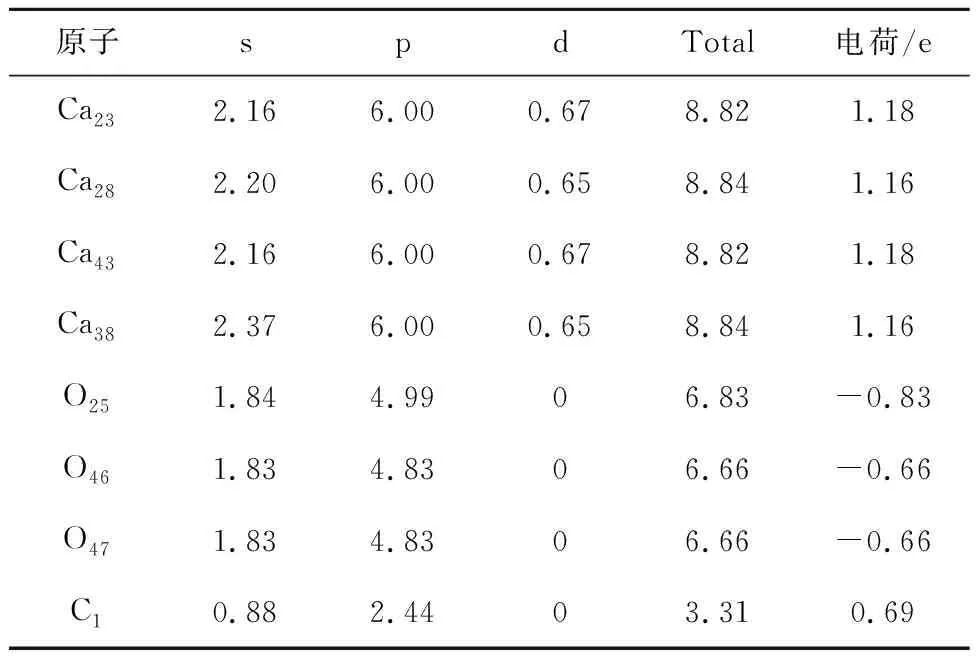
表13 CaO对CO2吸附过程Mulliken电荷分布情况Table 13 Mulliken charge distribution during the adsorption of CO2 by CaO
3 金属氧化物耦合含氮生物质炭吸附CO2
依据上述计算、分析结果,选择含N-X基团生物质炭与CaO进行耦合。MA等[26]采用DFT研究了在钙环化条件下,CaO/Ca12Al14O33吸附剂对CO2吸附机理;WONGTHONG等[33]构建了氧化镁/多孔碳体系,研究唑来膦酸(ZA)在氧化镁改性多孔碳表面吸附机理;信晶[34]在模拟分析CaO对焦炭表面NO还原作用机理时,构建了CaO协同焦炭体系表面NO吸附及还原路径。综上,构建CaO耦合含氮生物质炭体系,研究CaO与含吡啶氮氧化物生物质炭耦合作用下对CO2的吸附特性。首先对CaO重新构建(图10,长、宽、高分别为1.708 22、1.708 22、1.983 16 nm),然后在GGA-PBE泛函下进行几何优化,最后进行CO2吸附,收敛参数同第1节(CaO切面大于生物质炭边缘模型考虑排除CaO切面与生物质炭相对位置对协同作用下吸附的影响)。
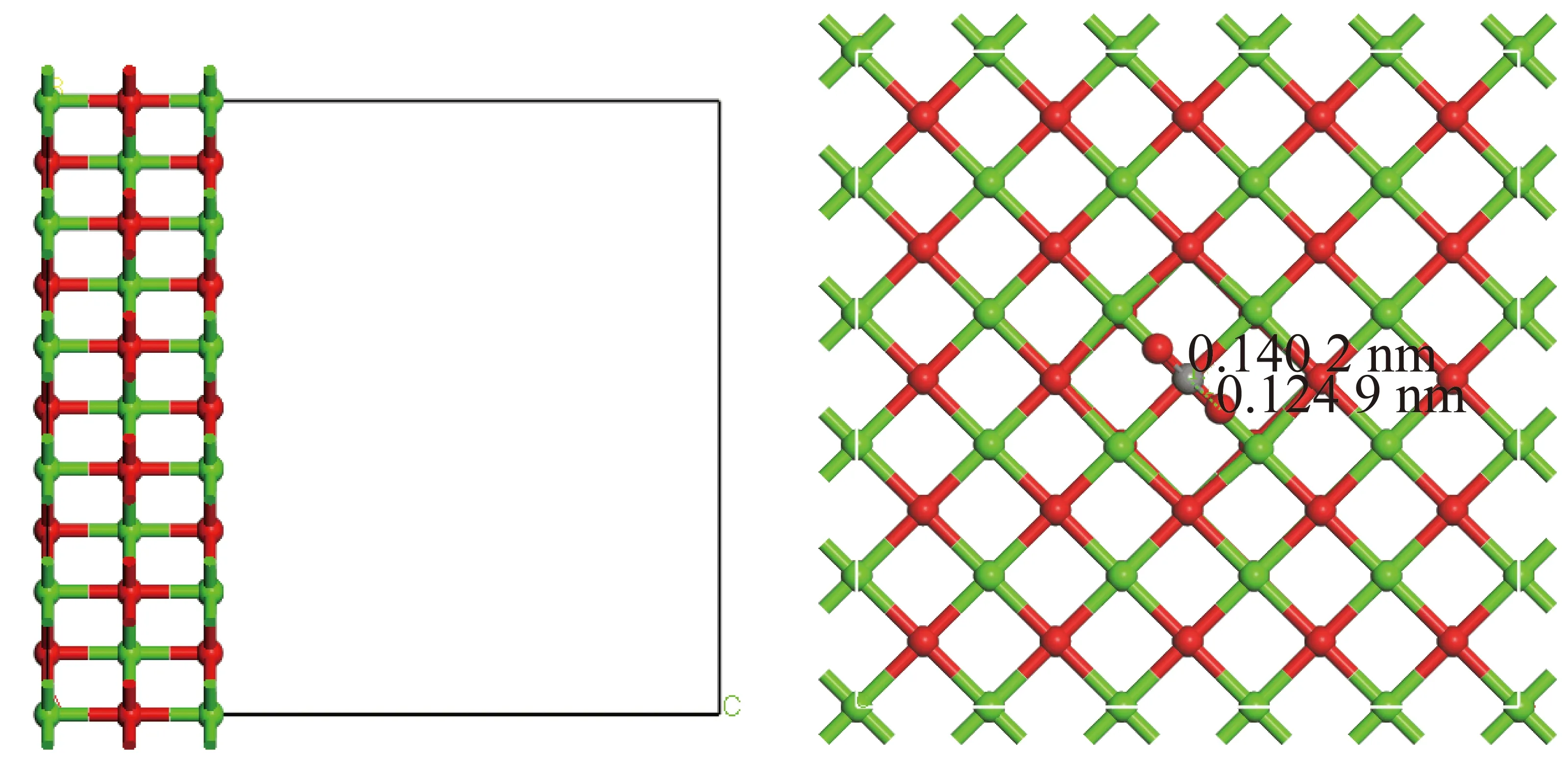
图10 新构建CaO及其吸附CO2Fig.10 CaO and its adsorption of CO2
根据CO2与生物质炭的相对位置,对上、平行、下3种位置进行CO2吸附。其中平行、下的吸附结构均与CaO单独吸附CO2保持一致的O-Top结构。CO2在CaO耦合含N-X基团生物质炭上不同吸附构型如图11所示,其吸附能见表14。
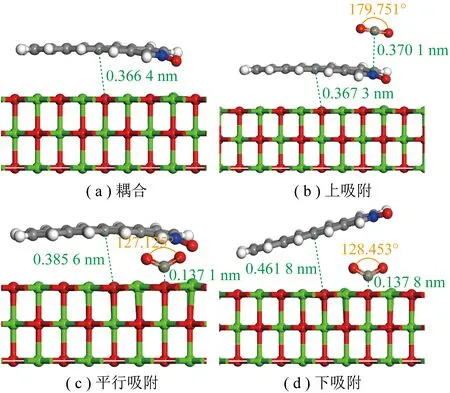
图11 CO2在CaO耦合含N-X基团生物质炭上的吸附Fig.11 CO2 adsorbed on CaO-coupled N-X group biochar
由表14可知,CaO与含N-X生物质炭耦合后上吸附和下吸附分别比CaO单独吸附CO2的吸附能高-137.40和-24.12 kJ/mol。而其在平行吸附方式下对CO2的吸附能比CaO单独吸附CO2吸附能高-0.87 kJ/mol,比CO2在含N-X生物质炭平行吸附方式下吸附(表3,-4.72 kJ/mol)提升-136.83 kJ/mol,确定CaO与含吡咯氮氧化物基团生物质炭协同作用下不同吸附位置影响吸附效果。与生物质炭吸附CO2相比,CaO掺杂后存在协同效应。掺氮生物质炭协同金属氧化物平行吸附下对CO2吸附效果较显著,原因可能为平行吸附除共价键作用外,还存在范德华力、氢键等弱相互作用力。
4 结 论
1)相较含N-5、N-6和G-N的生物焦,含N-X生物质炭对CO2吸附影响较大,与CO2间存在弱相互作用力。
2)CaO的O-Top位点对CO2吸附效果好,CaO中O较MgO对CO2中C作用更强,电子云重叠区域更大、电荷转移更多,同时Ca较Mg更易失去电子。
3)含N-X生物质炭协同CaO对CO2吸附有协同促进作用。与含N-X生物质炭吸附CO2相比,吸附能提升-136.81 kJ/mol;与CaO吸附CO2相比,吸附能提升-0.87 kJ/mol,原因可能是存在共价键的同时,还存在范德华力、氢键等弱相互作用力。
