超临界水氛围下萘开环机理
2023-10-20张英佳黄佐华
赵 浩,张英佳,黄佐华
(西安交通大学 动力工程多相流国家重点实验室,陕西 西安 710049)
0 引 言
煤炭超临界水气化技术是一种新型的煤气化技术,利用超临界水的强氧化性和高溶解性,将煤直接转化为H2、CO、CO2等产物,同时将煤中氮、硫、重金属等污染物以固体无机盐的形式沉淀,是实现煤高效清洁低碳利用的理想技术之一[1-3]。近年来针对超临界水煤气化过程的试验研究很多,主要集中在分析温度[1-4]、压力[5-7]、流量和催化剂等系统[8-11]运行参数对气化效率的影响。研究表明,温度是超临界水煤气化转化的决定因素,在中低温条件下超临界水煤转化过程中难以完全气化,导致反应器堵塞[12]。虽然提高反应温度能保证煤完全转化,但会严重腐蚀反应器并造成大量不可逆热损失。实现中低温条件下煤的完全转化仍存在巨大挑战。超临界水煤气化过程十分复杂,涉及水解、热解、脱水、加氢、聚合和异构化等一系列反应[13],同时由于超临界水(温度大于374 ℃、压力高于22.1 MPa)的特殊环境,限制了试验装置在线测量,导致超临界水煤气化过程的动力学机制尚未明晰。
多环芳烃的开环反应被认为是超临界水煤气化过程关键速控步反应,目前文献中普遍认为超临界水是芳香环开环的促进剂[13],但具体反应机制尚不清楚,对此主要有2种不同解释。JIN等[14]采用反应分子动力学方法结合密度泛函理论对煤替代物蒽在超临界水系统中的气化过程进行模拟,比较了蒽直接热解、亚临界水蒸气氛围及超临界水氛围中气化过程的差异。研究发现,超临界水系统中氢产率远高于热解和水蒸气气化系统,这与试验得出的规律一致。轨迹分析显示,超临界水通过与萘发生加成反应形成复合物,使C—C键裂解能由776.7 kJ/mol 降至218.5 kJ/mol,加速了芳香族化合物的开环过程。HAN等[15]采用反应分子动力学方法对煤替代物萘在超临界水系统中的气化过程进行模拟,研究指出,超临界水不仅提供了生成H2的氢原子,同时还提供了生成CO中的氧原子。在开环过程中,超临界水首先通过提供OH自由基或直接与萘发生脱氢反应生成萘自由基,随后通过分子碰撞促进开环过程。
由于模拟结果包含上万种组分信息和基元反应,缺乏反应动力学层次的分析技术,导致目前主要通过人工发现关键反应,且仅能获得定性水平的结论,使超临界水煤气化反应的关键速控步机理仍存在争议。在现有分析程序中,美国桑迪亚国家实验室开发的开源LAMMPS程序[16]中REAXC模块[17]能输出模拟过程中系统化学键和物种变化,但难以提供任何化学反应层面的信息。DÖNTGEN等[18-19]基于Python提出了Chemtrayzer开源程序,该程序通过分析反应过程的化学键变化给出了详细的化学反应路径,但计算效率较低,仅适用于小分子体系。中国科学院过程工程研究所李晓霞等针对煤体系提出了VARxMD程序[20],该程序在对3D化学结构的唯一物种识别、反应位点识别、化学键识别的基础上,能对模拟结果进行可视化分析,并生成完整的化学反应列表,该软件非开源,代码并未公开。华东师范大学朱通等开发了ReacNetGenerator开源程序[21],该程序可从反应轨迹中自动提取反应网络,并成功预测了RP-3替代模型燃烧过程的反应轨迹。
因此,笔者构建针对反应分子动力学模拟结果的化学反应路径分析方法,用以分析超临界水氛围多环芳烃详细反应过程,揭示关键自由基的形成和消耗途径,并确定开环反应的关键反应路径;利用定量化评估方法对比不同工况结果,实现分析结论的低温外推,为超临界水多环芳烃的动力学机理建模和模型优化提供依据。
1 计算方法
1.1 模拟方法
分子动力学方法结合反应力场是多相体系化学反应模拟的主要手段,宾夕法尼亚大学VAN DUIN等[22]提出了反应力场方法,该方法采用键级描述系统中原子间化学键强弱,键级由原子之间的距离和原子对应的化学价决定。键级能动态描述系统键能、键角能和二面角能的变化,因此可表征系统中化学键的断裂和生成。此外,反应力场用电负性平衡方法处理极化作用,考虑了范德华和库伦相互作用,参数化拟合后的反应力场能较好重现密度泛函方法精度下的反应势能面。相比高计算成本的第一性原理模拟方法,反应力场方法计算效率可提升3个数量级以上[22],实现数十万原子在纳米尺度下和纳秒时间内模拟,并在原子尺度下获得系统反应过程中的温度分布、压力分布、物种浓度和化学反应速率等基本参数,对吸附、扩散和相变等多相反应过程进行准确描述[23]。分子动力学结合反应力场的方法是一种原子间反应势驱动的方法,仅需构建反应力场信息而无需预设反应路径,这是其应用于复杂体系微观化学反应机制研究的重要优势。选取ASHRAF等[24]提出的CHO-2016力场势函数参数,相比2008年的CHO-2008参数,显著改进了小分子烃类和水的热力学和动力学描述精度,是目前针对烃类分子动力学模拟最准确的力场参数集。各研究案例的详细参数见表1,水的化学计量系数通过假定萘与水反应完全生成H2和CO2来确定,数值为20。
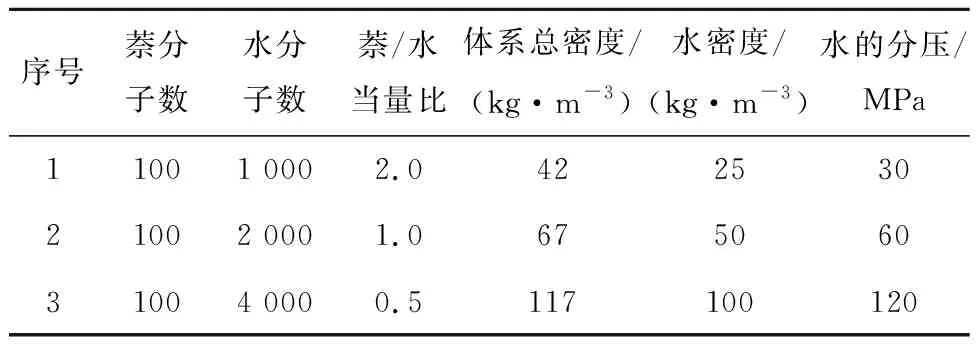
表1 各模拟案例的详细参数Table 1 Parameters for each simulation case
对比了400与100个萘分子系统的净反应发生次数统计(表2),可知2个系统差异较小。考虑计算耗时,选取100个萘分子作为模拟参数,并对所有模拟案例进行4次模拟取平均,以减少系综误差。利用Packmol软件[25]将萘分子和不同数目的水分子在空间中随机布置完成初始模型构建,周期性边界条件立方体盒大小为10.6 nm×10.6 nm×10.6 nm,保证所有模拟工况中水分压大于水的临界压力22.1 MPa,保证水处于超临界状态。模拟系统中固定萘浓度不变,通过调整水分子数实现当量比变化。
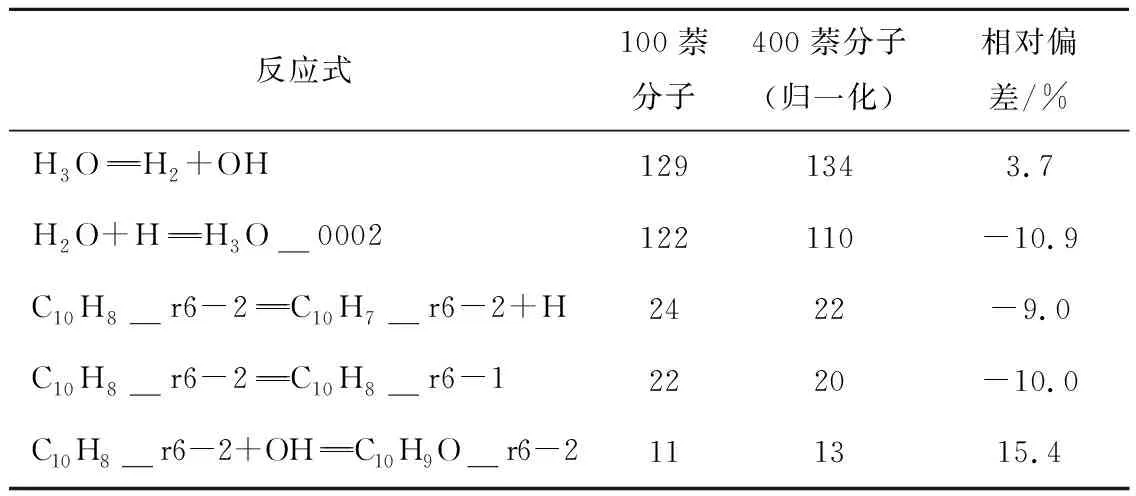
表2 不同分子系统部分净反应发生次数比较Table 2 Comparison of net reactions times in different systems
受限于计算机性能,反应分子动力学模拟的最大时间为纳秒数量级,为保证研究系统充分反应,首先在2 000~3 000 K进行模拟,通过追踪萘分子浓度,发现2 500 K下萘分子在8 ns内完全消耗,因此,选取2 500 K作为反应的最低模拟温度。此外,在2 600和2 700 K下进行分子动力学模拟,通过对比不同温度的模拟结果,对低温工况进行外推。选取0.1 fs作为时间步长避免数值误差累积,选用恒定数目、恒定体积和恒定温度的NVT系综进行模拟,温度阻尼为时间步长的200倍即20 fs。模拟过程中,将系统进行能量最小化优化,消除初始结构中不合理的原子位置,再对所有原子分配满足温度为1 000 K的高斯分布初始速度,并在1 000 K下进行10 ps初始平衡,200 ps内将系统温度升至目标模拟温度,在目标温度下固定温度进行模拟并输出系统模拟结果,结果分析中的零时刻均指升温完成时刻。所有模拟均通过开源分子动力学模拟软件LAMMPS[16]完成。
1.2 分析方法
键级是描述模拟系统反应过程化学信息的重要参数,不仅用于识别物种的唯一结构,还可用于描述基元反应变化。在Chemtrayzer基础上,结合图论分析库重现,从模拟系统中的键级进行物质识别和基元反应识别,并对基元反应进行统计获取反应路径,同时利用并行计算技术提高分析速度,运行流程如图1所示。程序首先读取LAMMPS输出的键级数据,并将键级进行二值化处理,即键级大于阈值0.3时则认为2个原子间存在化学键。需要说明的是,本研究仅关注芳香环断键过程,不考虑化学键是否为单键、双键等键类型信息,忽略键类型信息可减少同分异构体带来的反应路径噪声,同时可不丢失芳香环的结构信息。根据键级通过拓扑学理论进行连接性搜索,寻找与某原子存在连接关系的所有原子,并调用Open Babel程序库[26]将连接关系与各原子对应元素类型进行识别,转换成SMILES字符串[27-28],同时利用Open Babel程序SSSR模块分析结构的环特性,实现某一时刻下系统化学信息分析。从时间维度分析,如果不同时刻下某一原子的化学键信息发生变化,则根据化学键的生成或断裂情况和对应发生反应前后时刻的物种,生成对应的基元反应化学式。通过区分和合并正、逆化学反应式处理反应过程中大量逆向反应,获得整个反应过程各基元反应累积发生的次数统计,在此基础上进行反应动力学水平分析,获取从反应物到稳定产物的详细反应路径。此外,原始反应路径中存在大量仅活性位点不同的同分异构体,使反应路径稀疏,难以提取有用信息。为发现关键反应通道,分析过程中合并了分子式相同和碳环结构相同的物种。合并简化反应路径,如对于反应A+B<->C,当C物种的生成路径仅存在C<->D+E时,2个反应将合并为A+B<->D+E。考虑到模拟温度达2 700 K,高温下分子碰撞频繁,为保证精确捕捉单个化学键断裂的瞬间,使识别反应为仅涉及一个化学键断裂的基元反应,分析步长选取了比Chemtrayzer[18-19]建议值20 fs更低的1 fs分析步长,对应为模拟时间步长的10倍,通过合并正、逆化学反应式处理小步长带来的震荡反应。识别键级文件中时间戳信息,将键级文件分块,利用多核并行处理不同时刻的键级状态,提升分析效率,后处理耗时相较LAMMPS模拟耗时短1个数量级;将键级文件进行实时压缩,使磁盘占用减少一个数量级,解决更小分析步长导致的更多分析耗时和磁盘占用等问题。分析程序通过Python语言实现。
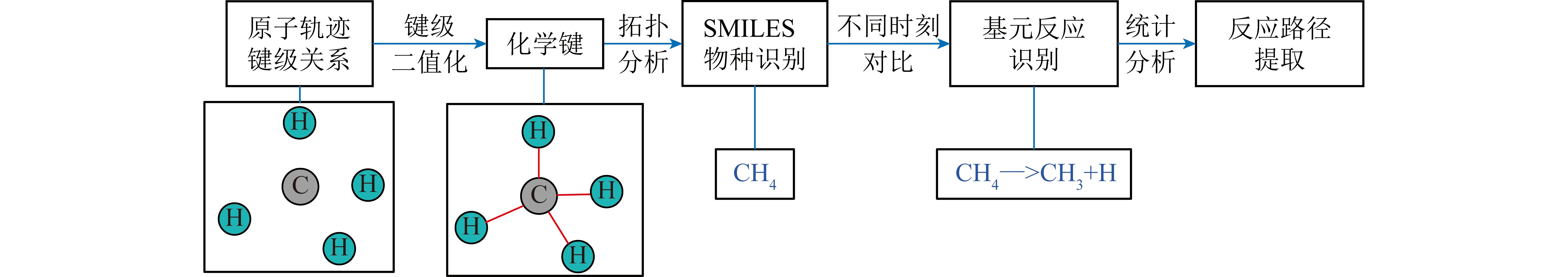
图1 后处理程序的分析流程Fig.1 Flowchart of the post-processing program
2 模拟结果分析
2.1 边界条件对反应过程的影响
2 500 K下当量比对反应物萘和中间产物CO的影响如图2所示(N0为初始值,φ为当量比,λ为衰减常数,α为拟合系数),模拟过程中固定萘分子浓度,通过控制水分子浓度调节当量比。可知当量比变化对萘分子消耗速率影响较小,说明萘分子消耗与水分子浓度呈弱相关,高温条件热解反应主导反应进程。水分子浓度对CO生成速率影响显著,由于反应系统中氧原子均来自水分子,说明水分子参与了CO生成过程,控制基元反应需进一步动力学分析。
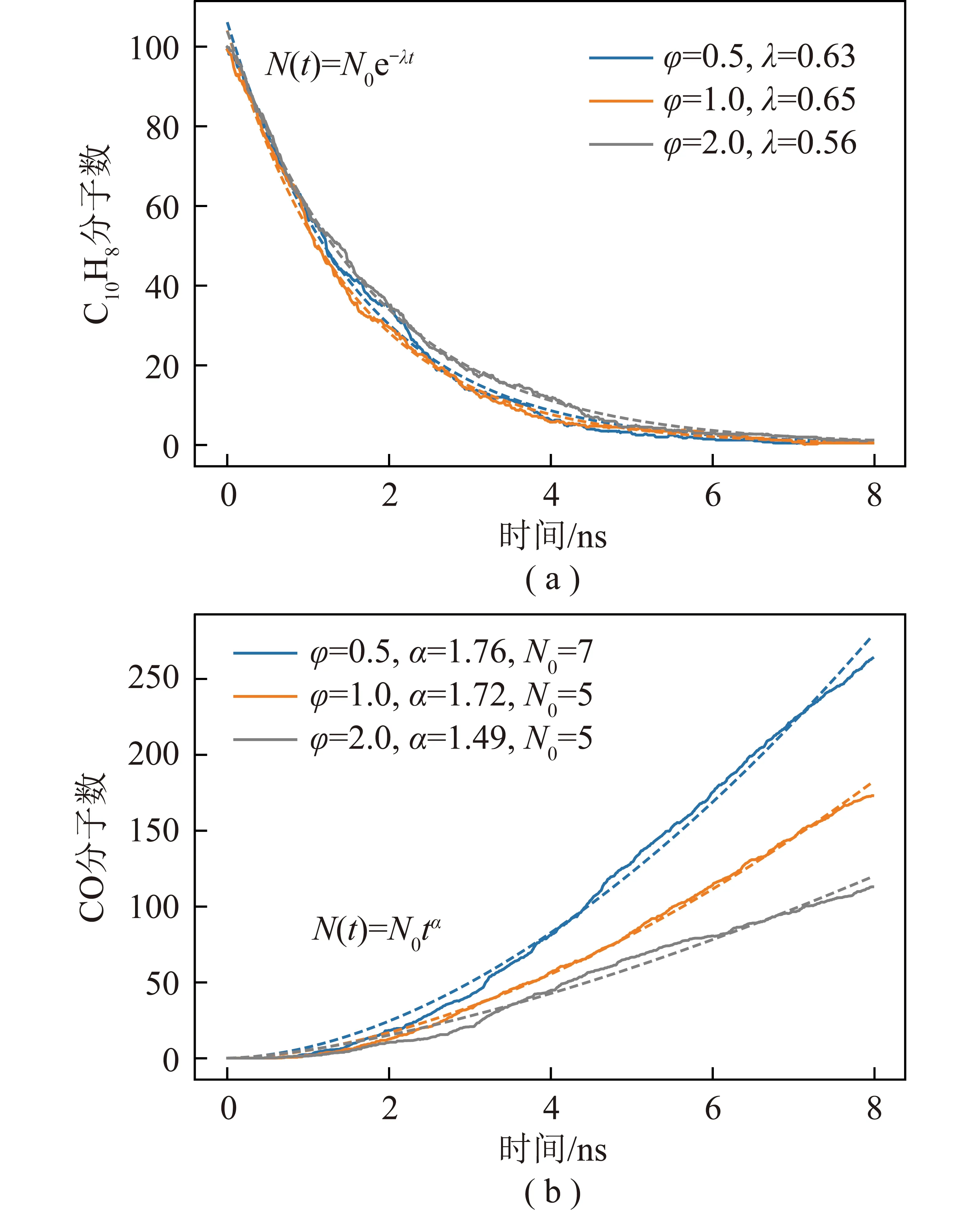
图2 2 500 K不同当量比萘和CO分子数随时间变化Fig.2 Molecular numbers of naphthalene and carbon monoxide with different equivalence ratios at 2 500 K
温度是影响反应快慢和反应路径变化的重要参数,因此,对比了温度对反应物浓度的影响如图3所示。可知反应物呈指数下降,且下降速率随温度升高而增加。曲线呈指数函数衰减,衰减常数λ与温度呈线性相关,衰减常数越大意味着反应物氧化耗时越长。反应物消耗95%时,模拟时间分别为4.58、3.02和1.58 ns,说明更低温度的反应分子动力学模拟耗时呈指数增长趋势。反应物浓度与指数函数较好的吻合行为说明当前模拟条件下反应过程速控步为一阶反应,该反应过程与温度强相关。
2.2 反应路径分析
为分析反应物萘的消耗路径,阐明萘开环反应路径主导的基元反应,对2 600 K、当量比为1条件,萘消耗95%时的反应路径进行分析,如图4所示,图中合并了同分异构体,不区分反应位点。可知:① 萘的消耗路径包括热解、脱氢、加成反应,其中热解是主要消耗路径,占比超过60%,热解反应的高占比与模拟温度直接相关;② 萘外部碳碳键比内部碳碳键更易断裂,萘外部碳碳键断裂消耗占比为内部断裂占比的5倍以上,但萘脱去一个氢原子后内部碳碳键断裂消耗占比与外部相当;③ 对于开环反应,即使在2 600 K下,萘分子脱氢反应仍为开环的关键步骤,占比超60%;④ 水分子难以直接与萘分子发生加成反应形成化学键,相比之下,萘分子脱氢后,水分子中的氧原子加成到萘原来的脱氢位点更容易,此后水分子再消除一个氢原子形成醇类,最后发生开环反应,这与JIN等[14]的结论不同。尽管本文后处理分析中捕捉到了萘分子与水分子的加成基元反应,但考虑其逆反应过程后,净反应发生次数在反应路径中占比不超过0.5%,故为非主要反应路径。

图4 2 600 K下当量比为1时萘消耗95%时的萘开环反应路径Fig.4 Ring opening reaction pathway diagram at 2 600 K with 95% naphthalene consumption at an equivalence ratio of 1
不同温度下直接消耗萘分子反应的发生次数见表3。由于反应过程中存在循环反应过程,且不同温度下模拟进行的时间不同,使净反应发生次数绝对值对比规律性不强。
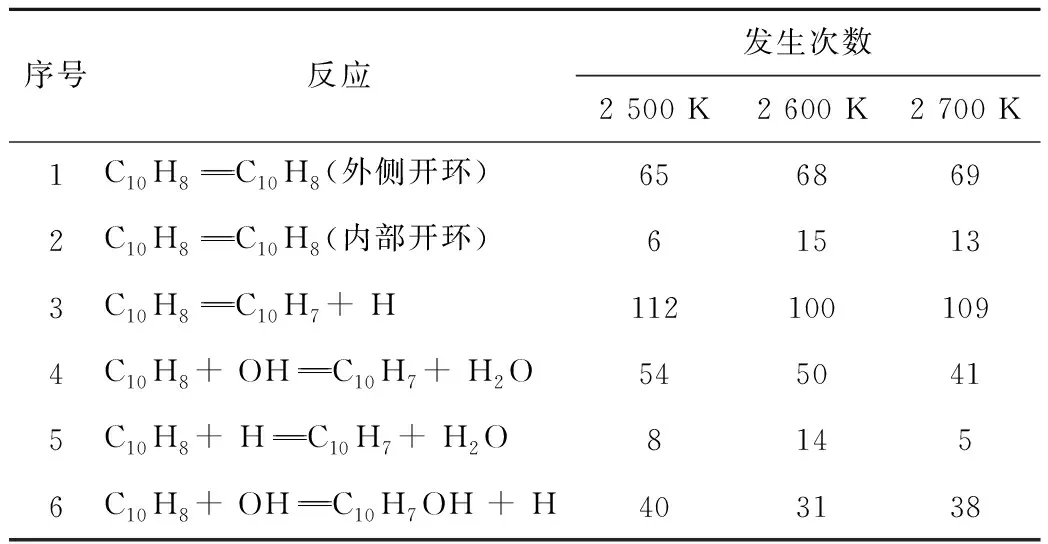
表3 当量比为1时不同温度下萘消耗95%时直接消耗萘分子净反应发生次数Table 3 Net reactions times that directly consume naphthalene with 95% naphthalene consumption at an equivalence ratio of 1
将数据进行归一化处理,根据反应类型定义反应1~3为热解主导反应类,反应4~6为自由基主导反应类。自由基反应类在萘分子总消耗比值与温度的关系如图5所示。可知随温度降低,自由基反应类占比不断提升,进一步通过线性拟合将结果进行低温外推发现,温度低于2 000 K时,自由基反应类占比超过50%,温度低于1 000 K时自由基反应类占比将超过75%。尽管实际超临界水煤反应过程中自由基相关反应主导反应过程,但该结论仍需试验和相关理论计算进一步验证。
2.3 物种通量分析

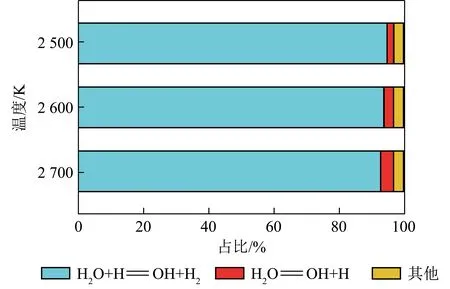
图6 当量比为1时不同温度下生成OH自由基的基元反应Fig.6 Main elementary reaction of OH radical formation at an equivalence ratio of 1 with different temperatures

图7 当量比为1、2 500 K下萘消耗95%时生成氢原子的主要基元反应Fig.7 Main elementary reaction of generating hydrogen atom when naphthalene consumption is 95% at an equivalence ratio of 1,2 500 K
3 结 论
1)采用反应分子动力学方法研究了不同温度和不同当量比条件下超临界水和萘的气化过程。计算结果表明,高温条件下萘分子数呈指数下降,消耗速率与当量比呈弱相关,呈现明显的一阶反应特性。通过分析系统每一时刻的键级变化,获得了定量化的基元反应路径,萘的开环反应路径包括直接热解、脱氢后开环和OH加成开环。
2)高温条件下直接热解占主导,1 000 K下脱氢和OH加成反应占主导。本研究与文献结论并不一致,超临界水直接提供活性H原子和OH自由基以及超临界水直接与反应物发生反应的占比极小,非主导反应。
