H2O2/Fe2(MoO4)3体系中H2O2吸附分解及NO氧化行为的DFT研究
2023-10-20王长安车得福李彩亭
刘 璇,吕 强,王长安,车得福,李彩亭
(1.湖南大学 环境科学与工程学院,湖南 长沙 410082;2.环境生物与控制教育部重点实验室(湖南大学),湖南 长沙 410082;3.西安交通大学 动力工程多相流国家重点实验室,陕西 西安 710049)
0 引 言
我国超过一半的SO2及NOx排放来自于电站及工业锅炉煤的燃烧。为控制硫、氮污染物的排放,燃煤电厂目前普遍采用石灰石-石膏湿法脱硫(WFGD)和选择性催化还原脱硝(SCR)2套系统分别脱除烟气中的SO2和NOx。这种分级串联式的治理模式虽然能实现单一污染物的高效脱除,但多套烟气净化设备的串联组合不可避免造成锅炉尾部烟气处理系统庞杂、占地面积过大、投资运行费用过高等问题。在煤电超低排放的大背景下,如何针对燃煤烟气硫、氮污染物开发经济高效的一体化脱除技术对于我国煤炭清洁高效利用、烟气污染物持续减排,深入打好蓝天保卫战均具有重要的战略及现实意义。

尽管通过试验方法可确定H2O2/Fe2(MoO4)3体系中氧化NO的主要活性氧物种,但在微观层面上,Fe2(MoO4)3催化剂具有高催化活性和高稳定性的内在原因、H2O2分子在Fe2(MoO4)3表面的吸附分解行为及体系中NO的氧化机制仍未清晰揭示。基于此,笔者拟借助密度泛函理论(DFT)计算的优势,首次计算研究H2O2和NO分子单独及二者同时在Fe2(MoO4)3(001)表面的吸附、分解及氧化行为,并通过考察吸附能、键长、Mulliken电荷及氧化路径等特性揭示H2O2催化分解、表面羟基形成及NO氧化的具体机制。相关研究结果有望深入理解H2O2/Fe2(MoO4)3体系中NO氧化机理,并为降低非均相类Fenton体系脱硝时的H2O2消耗,提升催化活性及稳定性提供理论依据。
1 计算方法与模型
1.1 计算方法
所有计算均基于DMol3量子化学计算程序包[6]完成。电子交换关联能项采用基于广义梯度近似的Perdew-Burke-Ernzerh(PBE)泛函处理[7-9]。内核电子采用有效核电势(Effective Core Potentials,ECP)方法处理,该方法相对于全电子的处理方式具有更高的计算效率且能很好兼顾计算精度[10]。数值基组采用双数值轨道基组结合轨道极化函数(Double-numeric Quality Pluspolarization,DNP)考虑,该基组计算氢原子时加入了p轨道的极化函数,能更准确处理体系中氢键[11]。过渡态计算采用线性同步转变-二次同步转变(LST/QST)方法[12],并通过频率计算确认过渡态的合理性。DFT计算过程中采用高精度的收敛标准,分别为:① 电子自洽循环2.72×10-5eV;② 最大受力0.54 eV/nm;③ 最大位移5.0×10-4nm;④ 能量2.72×10-4eV。
小分子在催化剂表面吸附过程或吸附分解过程的能量变化ΔE按下式计算:
ΔE=Emol+sur-(Emol+Esur),
(1)
式中,Emol+sur为吸附分子在催化剂表面吸附后或吸附分解后的能量,kJ/mol;Emol为吸附分子的能量,kJ/mol;Esur为催化剂表面的能量,kJ/mol。
1.2 计算模型
笔者前期研究发现共沉淀法及溶胶凝胶法所制备的Fe2(MoO4)3均属于单斜晶系[3,13],因此本文计算中构建了单斜晶Fe2(MoO4)3结构模型(图1(a))。该模型中,铁-氧八面体和钼-氧四面体2种结构单元通过共用顶点的氧原子而相互连接。经收敛性测试(图2),Fe2(MoO4)3晶胞几何优化采用的原子轨道截断半径为0.49 nm,布里渊区K点分布为1×2×1。几何优化后所得Fe2(MoO4)3的晶胞参数为:a=1.603 nm、b=0.948 nm、c=1.600 nm,α=90°、β=108.30°、γ=90°。该结果与文献中单斜晶Fe2(MoO4)3试验值吻合良好(a=1.574 nm、b=0.925 nm、c=1.570 nm,α=90°、β=109.12°、γ=90°)[14]。Fe2(MoO4)3晶体模型的模拟X射线衍射(XRD)图谱与试验得到的XRD图谱对比如图1(b)所示。可知Fe2(MoO4)3晶体模型的XRD图谱与试验结果吻合良好,表明本文构建的计算模型能准确反映Fe2(MoO4)3的晶体结构。

图2 原子轨道截断半径及K点的收敛性测试Fig.2 Convergence test of orbital cutoff parameter and K points
Fe2(MoO4)3表面吸附计算时,选择较易暴露的(001)面作为吸附表面[15],表面模型如图1(c)所示。该吸附表面由12层金属原子组成(5层Fe原子、7层Mo原子),其中底部5层金属原子(2层Fe原子、3层Mo原子)保持固定,上部7层金属原子设置为弛豫(3层Fe原子、4层Mo原子),吸附表面设置1.2 nm真空层以避免周期性镜像间的相互作用。此外,表面吸附计算过程中系统考虑了吸附分子在八面体铁原子位点(记为Feoct)、四面体钼原子位点(记为Motet)、O原子位点及桥位的吸附情况。
2 模拟结果与分析
2.1 H2O2在Fe2(MoO4)3(001)表面的吸附分解特性
H2O2在催化剂表面的吸附分解是体系中NO有效氧化的前提。因此,首先研究H2O2在Fe2(MoO4)3(001)表面的吸附分解特性。吸附计算中考虑了H2O2分子在不同吸附位点(Feoct位点、Motet位点、O位点及桥位)以及H2O2分子以不同放置方式(O—O键与表面平行或垂直)的吸附情况。各初始构型经几何结构优化后,最终得到4种稳定的吸附构型,如图3所示。由图3可以看出,其中构型A的吸附位点为Motet位点,H2O2分子初始平行放置;构型B的吸附位点为Feoct位点,H2O2分子初始平行放置;构型C的吸附位点为Mooct位点,H2O2分子初始垂直放置;构型D的吸附位点为Feoct位点,H2O2分子初始垂直放置。

图3 H2O2在Fe2(MoO4)3(001)表面的吸附构型Fig.3 Configurations of H2O2 adsorption on Fe2(MoO4)3(001) surface
构型A中H2O2分子O—O键被拉长至0.320 5 nm,表明O—O键断裂,并且断裂后生成的2个羟基分别与表面的Feoct原子和Motet原子成键。Fe—O键和Mo—O键的键长分别为0.189 3和0.193 8 nm。构型B与构型A类似,H2O2分子在催化剂表面吸附后分解为2个羟基,并且分别与表面的Feoct位点和Motet位点成键。而在构型C中,H2O2分解为O原子和游离的H2O分子,O原子吸附于表面Motet位点并且形成键长0.175 2 nm的Mo—O键,而游离的H2O则与Mo—O键中的O原子形成氢键。表面Mo—O键形成后,Mo原子明显向靠近吸附分子的方向移动,这表明Mo原子和O原子之间存在强烈的相互作用。在构型D中,H2O2分子分解为2个羟基,并且2个羟基同时吸附于表面的Feoct位点,所形成Fe—O键的键长分别为0.184 7和0.185 8 nm。
以上4种吸附构型的吸附能、键长参数及H2O2分子的Mulliken电荷分析见表1。构型A、B的吸附能较接近,分别为-312.27和-327.26 kJ/mol,构型C的吸附能略高于构型A、B,这是由于构型C中H2O2分解为O原子和H2O分子且伴随额外氢键的生成。而构型D中,H2O2分解生成的2个羟基同时吸附于催化表面的Feoct位点,H2O2中O—O键长在该吸附构型中变化最小。此外,根据H2O2分子在吸附前后的Mulliken电荷分析可知,4种构型形成过程中催化剂表面均向H2O2转移部分电子,即H2O2在催化分解过程中充当电子受体,这一结果与Haber-Weiss机理相吻合[16]。
为进一步分析H2O2分子吸附分解过程中与催化剂表面间的相互作用,构型B中表面Fe、Mo原子及与之相连羟基中O原子的投影态密度(PDOS)如图4所示。可知Fe原子的d轨道与相连羟基中O原子的p轨道在-5.27、-3.45、-2.82和-1.89 eV处重合,类似地,Mo原子的d轨道与O原子的p轨道在多个能级处相重合,表明在该构型中表面Fe、Mo原子与H2O2分解所得羟基之间存在强烈的相互作用,并最终通过金属原子d轨道与氧原子p轨道杂化形成稳定的Fe—O键和Mo—O键。

图4 构型B中表面Fe、Mo原子及相连接羟基中O原子的PDOSFig.4 PDOS of surface Fe, Mo atoms and the neighboring O atom of hydroxyl group in configuration B
2.2 NO在Fe2(MoO4)3(001)表面的吸附特性
NO在Fe2(MoO4)3(001)表面的吸附计算考虑了NO分子在不同吸附位点(Feoct位点、Motet位点、O位点及桥位)及NO分子以不同放置方式(O原子朝下或N原子朝下)的吸附情况,最终得到的2种稳定构型如图5所示。其中构型A的吸附位点为Motet位点,NO分子初始以N原子朝下放置;构型B的吸附位点为Feoct位点,NO分子初始以N原子朝下放置。2种构型的吸附能、键长参数及Mulliken电荷分析见表2。

表2 NO在Fe2(MoO4)3(001)表面吸附能、键长参数及Mulliken电荷分析Table 2 Adsorption energies, bond lengths and Mulliken population of NO on Fe2(MoO4)3(001) surface
构型A、B中NO分子均以N原子朝下的方式分别吸附于表面的Motet位点和Feoct位点,二者吸附能分别为-89.92和-62.03 kJ/mol,这表明NO在催化剂表面更加倾向于吸附在Mo原子位点。此外NO分子在Motet和Feoct位点吸附后,其N—O键的键长分别由0.116 6 nm拉长至0.117 6 nm和0.119 1 nm,NO分子吸附后被有效活化。吸附构型A中Mo、N原子以及吸附构型B中Fe、N原子的投影态密度如图6所示。由图6可以看出,构型A中Mo原子的d轨道同时与N原子s轨道和p轨道杂化成键,而构型B中Fe原子的d轨道主要与N原子的p轨道杂化成键。
2.3 H2O2和NO同时在Fe2(MoO4)3(001)表面吸附氧化特性
为探究NO在H2O2/Fe2(MoO4)3体系中的氧化机制,对H2O2和NO同时在Fe2(MoO4)3(001)表面的吸附反应行为进行研究。计算中考虑了Feoct、Motet、O及桥位4种吸附位点,且H2O2和NO分子分别以平行或垂直于吸附表面的初始构型放置。初始构型经几何结构优化后,最终得到2种稳定的构型,如图7所示。由图7可以看出,其中构型A的吸附位点为Motet位点,H2O2分子初始垂直放置,NO分子初始以N原子朝下放置;构型B的吸附位点为Feoct位点,H2O2分子初始垂直放置,NO分子初始以N原子朝下放置。2种构型的吸附能及H2O2和NO分子吸附前后的Mulliken电荷分析具体见表3。

表3 H2O2、NO同时在Fe2(MoO4)3(001)表面吸附构型的吸附能及Mulliken电荷分析Table 3 Adsorption energies and Mulliken population of configurations of NO and H2O2 co-adsorption on Fe2(MoO4)3(001) surface

图7 NO、H2O2分子同时在Fe2(MoO4)3(001)表面的吸附构型Fig.7 Configurations of NO and H2O2 co-adsorption on Fe2(MoO4)3(001) surface
构型A中,H2O2分解为2个羟基,且其中一个羟基吸附于表面的Motet位点,另一个羟基与NO结合生成游离的HNO2分子。HNO2分子和表面羟基之间通过氢键连接。为确认吸附构型A中生成的游离分子为HNO2,对比了构型A中游离分子的结构参数与HNO2分子结构参数的试验值(表4)。由表4可以看出,构型A中游离分子的键长、键角参数均与HNO2分子的试验值吻合良好[17],因此证实构型A中NO被氧化为HNO2。由表3的Mulliken电荷分析可以看出,H2O2和NO在催化剂表面共同吸附过程中,H2O2分子得电子而发生还原分解反应,NO分子失电子而被氧化,吸附反应过程如式(2)所示。构型A中表面Mo原子、H2O2中2个O原子及NO中N原子的投影态密度如图8所示。由图8可以看出,表面Mo原子的d轨道与H2O2中一个O原子(记为O1)的p轨道在多个能级处重合(图8(a)、8(b)),表明Mo的d轨道与O1的p轨道杂化形成Mo—O键;另一方面,N原子的p轨道和s轨道与H2O2中另一个O原子(记为O2)的p轨道在多个能级处重合,进一步证实了HNO2分子中N—O键的生成。

表4 计算所得HNO2分子(构型A)及NO2分子(构型B)结构参数与试验值对比Table 4 Comparison between the calculated structural parameters of HNO2 molecule (configuration A) and NO2 molecule (configuration B) and experimental values

图8 构型A中表面Mo原子、H2O2中O原子及NO中N原子的PDOSFig.8 PDOS of surface Mo atom, O atom in H2O2, and N atom in NO of configuration A

(2)
在构型B中,H2O2发生分解反应,最终形成了一个吸附于表面Feoct原子的H2O分子和游离的NO2分子,并且H2O分子和NO2分子之间通过氢键连接。表4中构型B中游离分子的键长、键角结构参数与NO2分子结构参数的试验值吻合良好[20],进一步确认了该构型中生成的游离分子即为NO2。同时,由表3的Mulliken电荷分析可以看出,构型B吸附反应过程中,H2O2分子得电子被还原,而NO分子失电子被氧化,总体反应过程如式(3)所示。
(3)
总体来看,由于构型A、B的吸附能接近,因此NO在H2O2/Fe2(MoO4)3体系中可同时氧化为NO2和HNO2,这与笔者前期研究结果一致[3,13]。此外,由于H2O2在与NO的竞争吸附中处于优势地位(H2O2在催化剂表面的吸附能远大于NO的吸附能),因此H2O2分子将优先吸附于Fe2(MoO4)3催化剂表面,进而导致NO氧化生成的HNO2和NO2分子处于外围(仅通过氢键与催化剂表面作用)。HNO2和NO2分子易克服氢键从催化剂表面脱离[17,20-21],这可能是笔者前期研究发现Fe2(MoO4)3催化剂具有良好稳定性,且使用后催化剂表面未发现硝酸盐沉积的主要原因。
为进一步阐明H2O2/Fe2(MoO4)3体系中NO的氧化路径,以H2O2分解产生的O基团作为氧化NO的活性物种,计算了NO氧化过程中的能垒图(图9)。由图9可知,NO在H2O2分解所产生的O基团作用下,仅需跨越32.42 kJ/mol能垒即可被氧化为NO2。
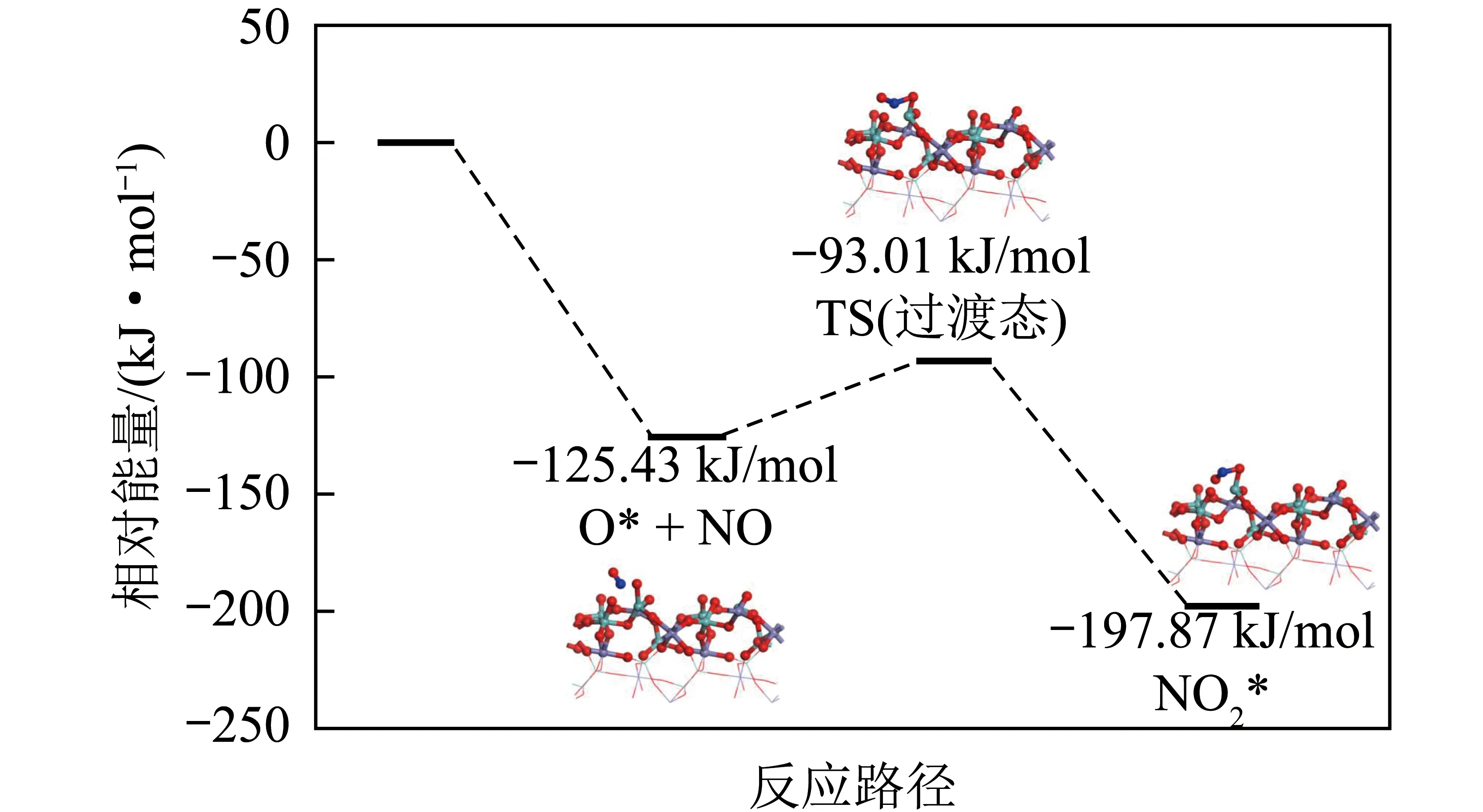
图9 H2O2/Fe2(MoO4)3体系中NO氧化的能垒图Fig.9 Reaction coordinate of NO oxidation in H2O2/Fe2(MoO4)3 system
3 结 论
1)H2O2在Fe2(MoO4)3(001)表面易催化分解为2个羟基,且有分解为表面O原子和游离H2O分子的趋势,所得稳定构型的吸附能为-247.81~-350.7 kJ/mol。Mulliken电荷分析表明H2O2吸附分解过程中电子由催化剂表面向H2O2分子转移,这与Haber-Weiss机理相吻合。
2)NO在Fe2(MoO4)3(001)表面上以分子形式吸附于Fe原子位点或Mo原子位点,但所形成稳定构型的吸附能(-62.03~-86.92 kJ/mol)均远小于H2O2在该表面的吸附能,表明在H2O2/Fe2(MoO4)3脱硝体系中,H2O2将优先吸附于催化剂表面,这有利于H2O2的催化分解及后续对烟气中NO的氧化。
3)H2O2和NO同时在Fe2(MoO4)3(001)表面的吸附计算表明,H2O2将优先吸附于催化剂表面并发生分解反应。H2O2均裂为2个羟基时,羟基中O原子的p轨道和NO分子中N原子的p轨道及s轨道杂化成键,最终生成游离的HNO2分子;类似地,当H2O2异裂为O原子和H2O分子时,O原子与NO成键最终生成游离的NO2分子。2种构型的吸附能大致相当,表明NO可在H2O2/Fe2(MoO4)3体系中可同时氧化为NO2和HNO2。
