多孔碳基非贵金属氧还原电催化剂研究进展
2023-10-19武鲁明于海斌王亚权
武鲁明,于海斌,王亚权
(1.天津大学化工学院,天津 300350;2.中海油天津化工研究设计院有限公司,天津 300131)
随着能源需求的不断增长和化石燃料资源的消耗加剧,可持续和可再生能源系统的转换和存储引起了人们的广泛关注[1]。燃料电池等可持续清洁能源的开发成为当前的研究热点。图1为典型的质子膜燃料电池[2],以氢气作燃料为例,氢气在阳极发生氧化反应,释放出质子和电子,质子穿过膜或分离器到达阴极,并在阴极和氧气发生还原反应。电子无法穿过聚合物膜从外部电路转移至阴极,当电子流过外部电路时,阴极和阳极之间存在电势差产生电流。具体的电极反应如下:

图1 氢/空气燃料电池的示意图[2]Fig.1 Schematic of hydrogen/air fuel cell[2]
阳极反应 H2→2H++2e-(Ea=0 V vs.SHE)
阴极反应 1/2O2+2H++2e-→H2O(Ec=1.23 V vs.SHE)
电池总反应 2H2+O2→2H2O(E=1.23 V vs.SHE)
标准电极电动势E为1.23 V,Ea、Ec和SHE分别表示阳极标准电动势、阴极标准电动势和标准氢电极。
阴极氧还原反应(ORR)是整个电化学反应的关键,其反应动力学缓慢,电极反应的发生需要克服一定的能量障碍,导致燃料电池的能量转换效率降低[3]。因此,需要寻找合适的催化剂提高反应动力,加快电池能量转换效率。目前高效铂基催化剂存在储量有限、价格高等缺点,造成质子交换膜燃料电池成本居高不下,阻碍了电化学能源技术的大规模商业化进程[4-5]。因此,开发高效、廉价的非贵金属ORR催化剂至关重要。
在已报道的非贵金属催化剂中,碳基非贵金属催化剂具有原料来源丰富、价格低廉、抗甲醇渗透能力强等优点,在酸性或碱性条件下的催化活性与Pt/C相当[6-7]。基于此,本文从非金属元素掺杂碳基催化剂、碳基非贵金属单原子催化剂和碳基非贵金属纳米颗粒催化剂3个方面出发(见图2),深入研究了当前碳基非贵金属催化剂的活性影响因素,总结了高效ORR 催化剂的调控机制,包括电子结构、活性位点密度与可接近性及调控碳/金属复合材料界面结构等,旨在为设计高效氧还原催化剂提供帮助,但碳基非贵金属催化剂稳定性有待进一步提升,高稳定性是其走向实际应用的重要指标。因此,本文进一步总结了催化剂在应用中的稳定性问题,并对电催化剂的发展前景进行了展望。

图2 多孔碳基ORR电催化剂分类示意图Fig.2 Schematic illustration of porous carbon-based ORR electrocatalysts
1 非金属元素掺杂碳基催化剂
碳材料具有独特的电子特性、可调的孔道结构、较大的比表面积及优异的导电性等优势,在ORR中表现出巨大的应用前景。氮、硫、硼和磷等杂原子掺杂可以显著增加碳材料的孔隙率,改善电极材料的传质性能和离子的可接近性。此外,基于杂原子和碳原子之间的电负性差异,杂原子掺杂可以调控碳材料的局部电子结构,打破碳材料电中性,改变氧分子的吸附状态,进而提高ORR 活性。图3 总结了非金属元素掺杂碳基催化剂的分类,主要包括氮元素掺杂碳基催化剂和多元素掺杂碳基催化剂两大类。
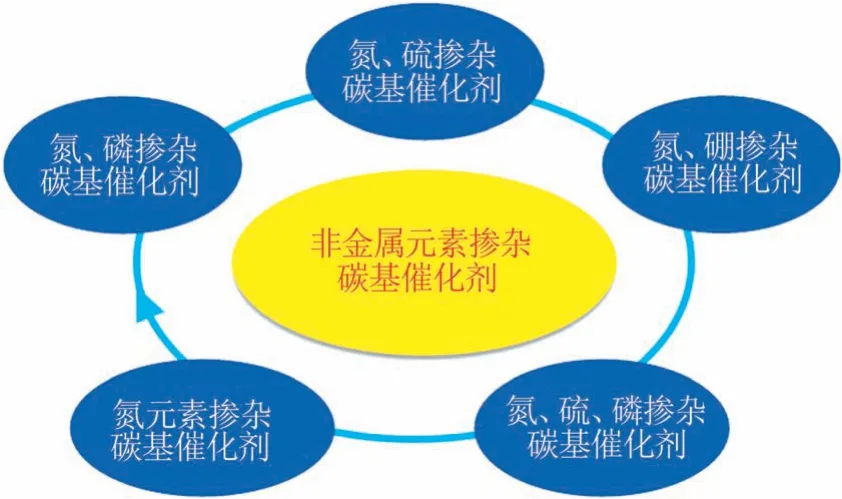
图3 非金属元素掺杂碳基ORR催化剂分类示意图Fig.3 Schematic illustration of carbon-based non-metallic ORR catalysts
1.1 氮元素掺杂碳基催化剂
氮原子的电负性大于碳原子,氮掺杂可以增加相邻碳原子的电荷密度,使活性碳位点数目增加,提升催化剂活性和稳定性。GONG 等[8]报道了一种氮掺杂碳纳米管垂直阵列,该催化剂在碱性条件下的活性优于Pt/C,同时具有抗CO 和抗甲醇毒化性能。计算结果表明,电子受体氮的掺杂使共轭碳平面上的原子呈现正电性,增加了相邻碳原子的电荷密度,加速氧气吸附的同时削弱了O—O 键键能,降低了催化剂决速步能垒,提升了ORR性能。
氮元素在六元环碳材料中的掺杂形式主要包括吡啶氮、吡咯氮和石墨氮[9]。吡啶氮具有一对孤对电子,可以提高碳材料的给电子能力,削弱O—O键键能,被认为是ORR 的主要活性来源;但也有学者认为石墨氮是ORR的主要活性来源。基于此,GUO等[10]设计了石墨模型催化剂——高定向热解石墨(HOPG),可控制备了4 种模型催化剂:吡啶氮掺杂HOPG、石墨氮掺杂HOPG、缺陷HOPG和HOPG。结果表明,吡啶氮掺杂的碳材料在酸性条件下具有最优的ORR 活性;吡啶氮使催化剂产生碱性位点,该位点位于吡啶氮邻近的碳原子上;ORR活性位点是吡啶氮毗邻的具有路易斯碱性的碳原子。
YANG 等[11]认为石墨氮是ORR 的活性中心,该团队制备了三维网状石墨烯纳米带(N-GRW)并探究氮掺杂类型对ORR 活性的影响,实验结果表明,供电子的石墨氮是ORR活性中心,三维纳米带结构促进了电子和电解质的转移和运输,N-GRW 展示出优异的ORR 性能。REN 等[12]通过热解脲醛树脂合成了氮掺杂的碳纳米球(NPCS-900),NPCS-900表现出优异的ORR活性,一方面归因于碳纳米片构建的微球具有较高的比表面积和多级孔结构,允许氮掺杂碳和缺陷碳活性位点最大化暴露;另一方面源于不同的氮构型(吡啶氮和石墨氮)在电催化活性中起着至关重要的作用。
1.2 多元素掺杂碳基催化剂
为了进一步提升催化活性,杂原子(如磷[13]、硫[14]、硼[15]等)掺杂碳材料被大量研究。多元素掺杂更有利于增强碳材料电荷转移能力,此外,掺杂元素之间的协同作用可以诱导碳材料形成更强的活性区域。对于氮、磷共掺杂碳材料而言,磷原子半径大于氮,给电子能力更强,磷掺杂可提升材料比表面积并产生结构缺陷,使碳材料具有较高的ORR 活性。ZHANG等[13]报道了一种三维氮、磷共掺杂的多孔碳材料,该多孔碳由聚苯胺和植酸形成的气凝胶经高温碳化得到。模拟计算结果表明氮、磷共掺杂效应和多孔碳结构对ORR活性至关重要。
硫原子电负性与碳接近,两者之间的电荷转移作用弱,但硫原子半径大于碳原子,硫原子掺杂使碳材料产生缺陷位,氮原子掺杂可以调节碳元素的电负性和配位环境,二者协同提升了碳材料在ORR中的催化活性。YANG 等[14]通过调控氮、硫掺杂类型及含量,合理地设计了一类在全pH 条件下具有ORR活性的催化剂(NSPCS)。表征结果表明,氮、硫共掺杂后,NSPCS 产生丰富的缺陷位,在ORR 中具有优异的催化性能。
硼原子电负性比碳低,其2p 空轨道缺电子,可夺取碳原子的电子使其具有更高的正电荷密度,氮、硼共掺杂可以有效增强电子转移作用。LU 等[15]制备了具有氮、硼共掺杂的石墨碳纳米笼,并从理论和实验两方面证实了NB-CN 独特的半开放纳米笼结构及富含缺陷的石墨碳结构可以增加电荷传输效率,并提供更多活性位点以提升ORR活性。
LONG等[16]采用三元素(例如氮、硫、磷)改性碳材料,研究了三元素掺杂碳材料对ORR 性能的影响。通过热解葡萄糖、三硫氰酸和磷酸制备的多孔碳材料可作为高效全pH范围ORR催化剂。该催化剂比表面积高达1 656 m2/g,在0.1 mol/L的氢氧化钠溶液中,半波电位高达0.862 V vs.RHE,极限电流密度为5.80 mA/cm2。
总的来说,非金属元素掺杂碳基催化剂具有价格低廉、分子水平结构可控和多种催化活性位点兼容的优点。通过杂原子掺杂可以调控碳基催化剂的电子结构和缺陷结构,使电荷再分配以优化碳基体,提高了碳材料的活性中心密度和催化活性,使其半波电位接近商用Pt/C的半波电位。但非金属元素掺杂碳基催化剂仍存在以下问题:1)在应用过程中,碳基催化剂的掺杂位点和缺陷位点易在高电势下被“腐蚀”,因而存在稳定性较差的问题;2)碳基催化剂的催化活性仍需进一步提升,以符合商业应用的要求;3)碳材料的高温热解过程较为复杂,精准地设计、合成具有特定掺杂位置和结构的活性位点仍具挑战性。因此,在碳基催化剂设计过程中,需要研究碳材料的腐蚀降解机制以提升其抗腐蚀性能。此外,需要结合理论计算和实验方法,辅助先进的原位表征技术,研究碳基催化剂的结构和组成变化,实现碳基催化剂的高效精准设计。
2 碳基非贵金属单原子催化剂
单原子(金属活性中心是分散在载体表面的孤立原子)M/N/C(M=Fe、Co、Ni、Mn、Zn等)催化剂因具有高原子利用率、低成本、优异的ORR活性等优点,被认为是铂基贵金属催化剂的高效替代品[17]。催化剂中非贵金属与氮配位形成的MNx活性中心具有高度分散的特性,降低成本的同时提高了金属原子利用率。研究表明,通过调控MNx活性中心的数量、电子结构及空间构型,能够提升催化剂的ORR活性和稳定性。图4 总结了碳基非贵金属单原子ORR催化剂的种类,主要包括铁基单原子催化剂、非铁基单原子催化剂和双金属单原子催化剂3类。
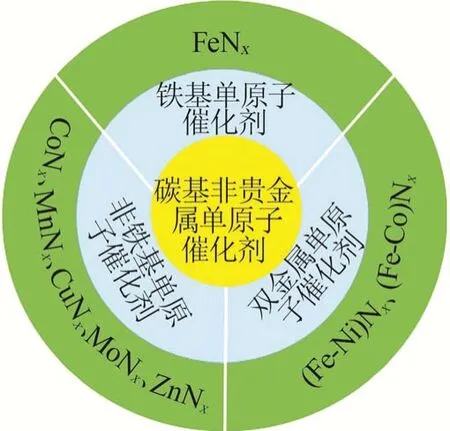
图4 碳基非贵金属单原子ORR催化剂分类示意图Fig.4 Schematic illustration of carbon-based non noble metal single-atom ORR catalysts
2.1 铁基单原子催化剂
M/N/C 催化剂的相关研究不计其数,铁基单原子催化剂的研究尤为突出,理论计算表明单个FeNx活性位点的活性与贵金属Pt 相当[18-19],从这个角度来说,M/N/C 材料具有与Pt 相当的活性。研究者常采用两种策略提升铁基单原子催化剂ORR活性:一是调控活性位点的电子结构增强催化剂的本征活性;二是提升原子活性中心数量和活性位点的可接近性。DODELET 课题组将高比表面碳载体BP-2000 与邻菲罗啉、醋酸亚铁充分球磨混合,进一步采用氨气高温处理,得到了Fe/N/C 催化剂。其中,氨气处理增加了碳材料的氮掺杂量和微孔数量,导致Fe/N/C催化剂活性中心密度增加[20]。
M/N/C 催化剂研究采用的碳材料为商业炭黑,其孔结构存在不易调控的缺点。ZIF-8(沸石咪唑酯骨架材料)因具有高孔隙率和高比表面积等优点,常被用作自牺牲模板制备单金属原子催化剂。DODELET团队后续采用ZIF-8代替商业炭黑,ZIF-8的高氮含量和丰富的微孔结构使催化剂的体积电流密度达到230 A/cm3,接近2015 年美国能源部设定的目标值(300 A/cm3)[21]。虽 然ZIF-8的微孔在ORR中起到担载活性位点的作用,但不利于反应物高效地传递至电极层。SHUI 等[22]通过调节聚丙烯腈和ZIF-8 添加量,调整了催化剂的孔结构和FeNx配比;在最优配比下,纳米碳纤维堆积形成了丰富的孔结构,使Fe/N/C 催化剂表现出优异的催化活性。CHUNG 等[23]采用聚苯胺、氰胺和金属盐制备了高孔隙率Fe/N/C 催化剂,采用像差校正扫描透射电子显微镜确定了FeN4的原子级图像,证实了FeN4是ORR的活性位点,对以后的研究具有重要的参考价值。
得益于ZIF-8配体的高氮含量,CHEN课题组采用在纳米限域条件下热解非晶相金属-配体配合物,得到多级孔金属/N/C 纳米球(Fe/N/C-HP)[24]。Fe/N/C-HP具有多级孔结构、高比表面积(1 389 m2/g)和高N掺杂量(原子数分数为9.4%),这些结构特征使Fe/N/C-HP在碱性和酸性介质中均展示出优异的ORR活性。邻菲罗啉配体可以和铁形成配合物,进而锚定更多的单原子铁,为进一步提升单原子铁掺杂量,该课题组采用纳米限域热解邻菲罗啉铁配合物,并用原位掺杂硫的方法制备多级孔碳纳米球(FeNx/NC-S)[25]。FeNx/NC-S 铁掺杂量高达3.7%(质量分数),在碱性条件下的ORR半波电位提升至0.92 V vs.RHE。
为明确单原子电催化剂的活性位,制备具有高稳定性的FeNx构型催化剂,XIA 等[26]采用以往未曾关注的高温条件(1 100~1 200 ℃)制备高性能Fe-N-C 催化剂,在此温度下热解的催化剂可有效去除产生过氧化氢副产物的非活性氮位点,并将稳定性低的D1结构(O-FeN4C12)转变为稳定性高的D2结构(FeN4C10),结构优化后的Fe-N-C 催化剂在PEMFC(质子交换膜燃料电池)中表现出优异的稳定性(H2/O2条件下运行30 h 性能保持率>80%),并且在35 d长周期运行过程中没有出现活性损失。
2.2 非铁基单原子催化剂
非铁基单原子催化剂(如CoNx、MnNx、CuNx、MoNx和ZnNx等)用于ORR,均具有优异的催化效果。例如,QU 等[27]研究了一种大规模合成Cu 单原子催化剂的方法,该方法利用NH3将块体铜金属氧化成易挥发的Cu(NH3)x组分,Cu被氮掺杂的多孔碳缺陷捕获,形成单原子Cu基催化剂。这种方法还可用于制备工业级镍、钴基单原子催化剂。
构建模型对探究原子级催化剂的活性具有十分重要的作用,调节原子配比和配位环境可实现单原子催化剂ORR 性能的提升。WANG 等[28]通过表面活性剂F123包覆金属有机骨架,热处理后得到具有核-壳结构的单原子Co-N-C@F127 催化剂,外层表面活性剂的限域效应抑制了Co原子的团聚,并抑制了ZIF 内部微孔结构的塌陷。密度泛函理论计算结果表明,原子分散的Co-N-C@F127 催化剂含大量的CoN2+2位点,这些位点具有高活性和热力学稳定性,有利于提升催化剂在酸性条件下的ORR活性。
锰基单原子和锌基单原子催化剂同样被应用于ORR 中,TIAN 等[29]通过设计螺旋状碳纳米管制备高度分散的Mn 原子位点,聚多巴胺修饰的螺旋状MnNC-PDA-700 催化剂具有大量MnN4活性位点,显示出优异的ORR 电催化性能,半波电位高达0.87 V vs.RHE,优异的电催化性能源于碳纳米管的螺旋结构和聚多巴胺分子在纳米管上的强包裹作用。DENG等[30]通过共热解ZIF-8、双氰胺和葡萄糖形成原子级分散的ZnNx碳材料(Zn-NC/GD),双氰胺和葡萄糖热解后在ZIF-8 表面形成一层碳壳,有效避免了高温下的Zn蒸气逃逸;Zn-NC/GD具有优异的半波电位(0.86 V vs.RHE)和较低的Tafel 斜率(97.8 mV/dec)。尽管上述文献表明锰基和锌基金属单原子催化剂具有丰富的活性位点和多孔结构,但其ORR活性低于铁基和钴基单原子催化剂,因此活性中心的选择和活性位的调控至关重要。
2.3 双金属单原子催化剂
双金属单原子催化剂是ORR的一个前沿研究,通过向单一过渡金属M-N-C 中引入第二组分金属元素,使相邻金属之间产生电子协同作用,调控活性位点的配位环境,进而表现出比单原子催化剂更优的ORR催化活性。其中,以Fe、Co为中心的M-N-C催化剂因具有最优的ORR活性被广泛研究。WANG等[31]构建了新型氮掺杂碳纳米管担载的Fe-Co双位点催化剂,精确地控制Fe3+前驱体与锌/钴双金属有机骨架中Co 位点之间的键合。客体金属铁与钴形成的铁-钴双金属位点吸附在双金属有机骨架的腔体内,高温碳化后双金属有机骨架转变为氮掺杂碳纳米管担载的双金属位点催化剂[(Fe,Co)/CNT]。ORR 测试结果表明,(Fe,Co)/CNT 的半波电位高达0.954 V vs.RHE,远高于Pt/C 电极的半波电位(0.842 V vs.RHE)。
为探究双金属单原子中心的催化机制,LIU等[32]提出了一种实现FeN4和CoN4双金属原子中心均匀嵌入N 掺杂石墨碳中的“预锚定金属孪晶”策略:Fe 和Co 的双核酞菁二聚体锚定在金属-有机框架(MOF)中,在热解前实现原位离域分散,得到在ORR反应中表现出优异电化学性能的FeCoDAC/NC催化剂。理论计算表明,相邻金属的协同作用优化了金属的d 带中心位置,平衡了*O 中间体的自由能,从而提高了ORR活性。
Ni、Fe双金属单原子催化剂的研究也取得了一定进展。CHEN 等[33]利用自组装法制备了Ni、Fe 双金属单原子催化剂Ni-N4/GHSs/Fe-N4,Ni 单原子位于中空碳纳米球的内壁,Fe单原子位于中空碳纳米球的外壁,镍和铁单原子与氮元素均是四配位结构;BAI 等[34]在ZIF-8 表面可控负载FeNi(mIm)x制备ZIF-8@FeNi(mIm)x前驱体,进一步可控转化为MOF衍生的原子分散的FeNiSAs/NC催化剂,其在碱性介质中ORR 半波电位较高(E1/2=0.91 V vs.RHE),密度泛函理论计算结果表明,羟基中间体在活性位点上的吸附能较低且ORR能垒较小,催化剂表现出较高的ORR活性。
总的来说,研究者通过调控金属中心原子的电子结构、配位环境和活性位密度及可接近性,提高了金属原子利用率和反应活性,使碳基单原子催化剂的ORR活性优于商业Pt/C,但ORR稳定性仍需进一步提升[35]。目前已提出的金属原子的团聚和流失、反应生成的H2O2对活性位点的氧化和腐蚀及微孔水浸等催化剂衰减机理将在第四部分进行总结。在催化剂的实际应用进程中,为了增强碳基单原子催化剂的稳定性,需借助原位表征技术等明确活性位点在ORR过程中的动态变化规律,以帮助理解反应机理和衰减机理,进而探究新的腐蚀降解机制,为制备高活性和高稳定性单原子催化剂提供坚实的理论指导。
3 碳基非贵金属纳米颗粒催化剂
在热解法制备ORR催化剂的过程中,过渡金属前驱体易在高温下团聚成金属纳米颗粒,与此同时杂原子掺入碳材料中形成缺陷碳。一方面碳基质的结构和组成对催化性能影响较大,例如,碳基质的多孔结构会影响电子传输及活性位点的暴露;另一方面纳米颗粒通过与外层包覆的碳层或者周围碳基质中的活性位形成协同作用促进ORR的进行。因此,杂原子掺杂碳材料和金属基催化剂的复合可以实现电催化活性和稳定性的提升,此外金属纳米粒子的种类、大小和分布影响其电催化性能(图5),碳基质的孔结构和形貌也是影响催化剂活性的重要因素。

图5 碳基非贵金属纳米颗粒催化剂分类示意图Fig.5 Schematic illustration of carbon-based non noble metal nanoparticles ORR catalysts
3.1 铁、钴基纳米颗粒/碳复合催化剂
碳材料因具有良好的导电性、较大的比表面积、优异的稳定性和抗腐蚀性能,常与金属铁、钴基纳米颗粒复合用于高效电催化反应。铁、钴基金属纳米颗粒催化剂的制备通常采用MOFs或金属配合物作为前驱体,在此基础上引入杂原子使催化剂产生缺陷位,形成更多的活性位,以提高ORR活性,但研究认为,铁、钴基金属纳米颗粒催化剂中不可避免地存在Fe/Co-N 位点,对这类催化剂催化机制的认识仍存在不足。
为探究ORR 活性中心,VARNELL 等[36]采用Cl2和H2处理含碳包裹的铁基纳米颗粒和FeNx两种位点的催化剂。Cl2处理后,催化活性中心中毒导致催化剂失活;进一步用H2处理,铁被H2还原得到碳包裹的铁基纳米颗粒。催化剂表现出与初始态相同的ORR活性,通过这种钝化和活化的方法明确酸性条件下催化剂的活性来源是碳包裹的铁基纳米颗粒,而不是通常认为的FeNx位点。
外层石墨碳与钴纳米颗粒之间的协同作用可以促进ORR的发生。LIU等[37]分别以二氧化硅胶体和聚氧化乙烯-环氧丙烷-环氧乙烷作为硬模板和软模板,制备了Co@NCNTs-800 催化剂,石墨碳层和钴纳米颗粒之间的协同效应表现为钴纳米颗粒可以激活石墨碳层,有利于ORR 反应的电子转移,进而增加ORR活性。此外,石墨碳层还可以保护钴纳米颗粒免受团聚和电解质腐蚀。
金属有机骨架ZIF-67因具有比表面积大、孔结构丰富且金属位点均匀分散等优点常被用作前驱体制备钴基催化剂。例如,GAO等[38]以硝酸钴和2-甲基咪唑为原料、聚乙烯吡咯烷酮为络合剂、碳纳米管(CNT)为添加剂,一步热处理制备N-Co@C/CNT 催化剂;ZIF-67 热解后得到的钴纳米颗粒较为均匀,CNT增加了N-Co@C活性位点的暴露程度和导电性。WANG 等[39]采用微通道反应器将ZIF-67 的合成时间缩短到毫秒级,制备了ZIF-67+聚乙烯亚胺(FEI)复合物衍生的钴纳米颗粒催化剂[Co@N-C(FNP)];与直接混合法制备的Co@N-C(DM)催化剂相比,微通道反应热解得到的Co@N-C(FNP)催化剂具有更高的Co含量,因此具有更多的ORR活性位点。
3.2 碳化铁纳米颗粒/碳复合催化剂
对于封装型碳化铁颗粒催化剂,提高封装型碳化铁纳米颗粒的密度或引入杂原子修饰催化剂结构,有利于设计出高活性的电催化剂。HU 等[40]采用一步热解法制备新型石墨碳均匀包裹的碳化铁纳米粒子,空心碳纳米球表面几乎没有杂原子氮或金属的掺杂。在酸性介质中,外部石墨碳层可以稳定碳化物纳米粒子,并活化外层的石墨碳层,使外部碳层具有ORR 活性。催化剂在酸性和碱性电解质中均具有很高的活性和优异的稳定性。
石墨碳包裹的铁基纳米颗粒被认为是高活性ORR 电催化剂,但是,复杂的金属组分及其在催化反应中的作用机制尚不清楚。为研究封装型催化剂活性中心,ZHONG等[41]制备了一系列形貌基本相同但组成不同的石墨碳层包裹的碳化铁颗粒催化剂(G@FeNPs)。由57Fe-Mössbauer 光谱鉴定得出,铁基催化剂组成包括α-Fe、γ-Fe、γ-Fe2O3、Fe3C及Fe3+高自旋态/Fe2+低自旋态引起的少量双峰组分。测试结果表明,ORR 活性与Fe3C 的含量呈正相关,证实了石墨碳层包裹的Fe3C纳米颗粒是ORR活性来源。
封装型碳化铁纳米颗粒的密度和分散度同样影响催化剂的ORR 性能。XIA 等[42]制备了中空纳米纤维封装的高分散碳化铁纳米颗粒催化剂(Fe3C@MHNF),其具有高电导率、多反应通道和高度分散的碳化铁活性中心,半波电位为0.90 V vs.RHE,超过了商用Pt/C 催化剂。GANGADHARAN等[43]采用三聚氰胺泡沫原位生长聚多巴胺的方法制备了3D-FePDC 催化剂;得益于丰富的碳包覆碳化铁中心和三维网络结构,3D-FePDC 在酸性和碱性条件下均表现出较好的ORR活性。
3.3 金属磷化物/碳复合催化剂
金属磷化物因具有优异的导电性和良好的耐酸碱性备受关注,磷化物中具有空d 轨道的金属原子与碳材料复合后,一方面两者之间发生电子相互转移作用,提高了催化剂的稳定性;另一方面,通过调整碳材料的配位环境,为ORR 中间体提供吸附位点,提升氧化还原反应动力学。催化剂组分、结构及表界面性质极大地影响了ORR 活性。研究者常采用元素掺杂、结构设计及调控碳材料与金属磷化物的界面相互作用等方法提升催化剂的ORR活性。
根据已报道的研究,铁基催化剂一般比钴基催化剂表现出更好的ORR 催化活性。NOROUZI 等[44]采用三苯基膦和氯化亚铁为前驱体,通过一步简单的热解制备了多孔磷和铁掺杂的碳材料(PFeC),PFeC具有丰富的Fe2P活性中心,在碱性和酸性介质中对ORR均具有电化学活性,半波电位高达0.88 V vs.RHE。PFeC 催化剂的优异电催化性能归因于P 和Fe原子之间的协同效应,Fe2P纳米颗粒均匀分布在碳基质上,提供了更多的活性位点,杂原子掺杂引起碳的电中性共轭结构被破坏,使得催化剂电导率提升,最终表现出优异的电催化活性。
为了探究石墨碳层和磷化铁之间的界面相互作用,NI 等[45]设计了一步法热解三聚氰胺、磷酸氢二铵和氯化血红素混合物,制备了氮、磷共掺杂石墨碳层封装的FeP 纳米颗粒复合催化剂(FeP@PGL),经表征发现,磷化铁纳米颗粒均匀分散在氮掺杂的碳基质上,外层磷丰富的石墨碳层和内部磷化铁之间存在电子相互作用,内部的磷化铁向外层石墨碳转移电子,引起电荷在界面上的重新分布,降低了催化剂表面的功函数,这种界面相互作用大大提高了ORR 活性。此外,外部石墨碳层可以保护FeP 免受氧化和溶解,FeP@PGL表现出优异的稳定性。
碳基非贵金属纳米颗粒催化剂的高电催化活性主要归因于:1)有利于电子传输及活性位点暴露的碳基质多孔结构;2)内部纳米颗粒向外层石墨碳转移电子引起电荷在界面上重新分布的碳/金属纳米颗粒间的界面相互作用;3)金属纳米颗粒、杂原子掺杂碳和可能的Fe-Nx-C 等多活性位之间的协同作用。目前,只有少数催化剂在酸性条件下的ORR活性可与商业Pt/C 相比。因此,需要通过以下方法提升碳基非贵金属纳米颗粒催化剂在酸性条件下的ORR活性:1)减小纳米颗粒的尺寸并增加封装型纳米颗粒的负载量;2)将具有特定功能的分子用于前驱体,尝试封装不同金属类型的纳米颗粒,以获得石墨碳层封装的混合金属纳米颗粒,进而产生更多的界面相互作用。
表1总结了碱性条件下碳基非贵金属催化剂的ORR 性能。由表1 可知,对于碳基单原子催化剂而言,其在碱性条件下表现出优异的ORR 活性,ORR半波电位为0.83~0.954 V vs.RHE。双金属单原子催化剂表现出较优的催化活性,可能是由于引入第二组分金属元素后,相邻金属中心之间产生的电子协同作用调控了活性位点的配位环境,以及催化剂对氧物种吸附能力的改善。对于非金属元素掺杂碳基催化剂而言,其在碱性介质中的ORR性能与商用Pt/C 接近,多元素掺杂碳基催化剂ORR 活性优于单一氮掺杂碳基催化剂,说明多元素掺杂更加有利于增强碳材料电荷转移能力,进而诱导碳材料形成更强的活性区域。碳基非贵金属纳米颗粒催化剂中,铁基催化剂表现出较优的ORR活性,ORR性能取决于碳基质的多孔结构、活性位点密度和碳/金属界面间的电荷转移作用等因素。

表1 碳基非贵金属催化剂的ORR性能对比Table 1 Comparison of ORR performance of porous carbon-based non noble metal catalysts
4 铁基非贵金属催化剂稳定性研究
尽管碳基非贵金属催化剂在高活性方面已经取得了重大进展,但是它在酸性条件下的高稳定性仍具有挑战性。目前,稳定性方面的研究工作主要围绕M-N-C 催化剂展开,基于已报道的研究,M-N-C在酸性介质中的活性衰减主要包括4 种机制:1)碳腐蚀引起的脱金属作用[63-64];2)H2O2的氧化作用[65];3)活性位点的质子化[66];4)催化剂的微孔浸没[67]。根据SHAO等[68]的研究结果,Fe-N-C的失活主要源于碳腐蚀脱金属和H2O2的氧化作用。
M-N-C材料中的碳载体不仅起导电作用,更是活性中心的主体。碳氧化可能会破坏活性位点,造成金属中心的流失,同时降低催化剂的稳定性。CHOI 等[69]通过在线电感耦合等离子体质谱和微分电化学质谱研究了高性能FeNxCy模型催化剂的降解机理,结果表明:当电压低于0.7 V时,发生铁颗粒脱金属现象,但金属脱除对ORR 活性无不利影响;当电压高于0.9 V 时,发生碳氧化和腐蚀现象,破坏了FeNxCy活性位点,导致ORR 活性降低;在0.7~0.9 V电压窗口内,可避免Fe-N-C 催化剂在酸性介质中的损失。FERRANDON等[70]采用X射线吸收近边结构光谱和电感耦合等离子体质谱确定催化剂在不同电势和电解质中的铁损失,结果表明,Fe 存在流失现象,且流失速率和流失量取决于施加的电势和电解质类型:低电势下脱金属化率高,但对ORR 活性无不利影响;由于Fe-N-C 活性位点的破坏常伴随着碳腐蚀与氧化的发生,在较高电势下,催化剂活性损失程度较大。此外,在燃料电池实际应用中,碳被氧化后催化剂更具亲水性[71],易造成微孔水淹现象,影响传质效果从而造成电池的性能损失。
除了碳氧化和脱金属造成催化剂的稳定性降低,中间体H2O2的生成同样影响催化剂的稳定性。当催化剂以二电子路径进行ORR反应时,会产生中间体H2O2,H2O2与Fe-N-C 中心的铁发生反应,形成羟基自由基,该活性氧物种一方面会攻击碳基底,导致严重的碳腐蚀,降低稳定性;另一方面会攻击质子膜,破坏膜电极结构。文献[72]研究了质子膜燃料电池ORR 中Fe-N-C 催化剂的主要失活机制:H2O2并不直接影响催化剂的金属活性位点,而是氧化FeN4结构周围的碳,削弱Fe-N4-C 与O2的结合能力,从而降低活性位点的活性。因此,设计和制备具有高稳定、高活性的碳基非贵金属催化剂是一项意义重大同时又富有挑战性的工作。
5 展望
本文系统综述了多孔碳基非贵金属催化剂用于ORR的相关研究进展,虽然碳基非贵金属ORR催化剂具有较优的催化性能,但其在ORR反应中的实际应用仍面临诸多挑战。在催化剂的活性方面:第一,碳基催化剂活性位点的精准控制仍具有挑战性,比如对于铁基单原子催化剂而言,活性中心的电子构型和配位环境具有多样性,对ORR具有不同的催化效果;第二,对复合型催化剂的活性中心和反应机制的理解仍存在争议,理论研究和更先进的表征技术不够完善;第三,催化剂的金属负载量低,活性中心和活性位点数量有限,导致催化剂活性不足,此外,碳基催化剂的产率较低,难以满足工业层面规模化制备的要求;第四,酸性介质中,高活性、高稳定性的ORR催化剂的合成仍是难题,在酸性条件下的活性和稳定性超越铂碳催化剂仍具有挑战性。因此,研究人员仍需发掘更先进的表征技术,如原位X 射线吸收光谱、X 射线光电子能谱、红外光谱法等,跟踪反应中间态,探究活性位点和反应中间体的动态变化规律;仍需开展理论研究来探究活性中心结构和组成与催化剂ORR 性能之间的关系;最后,通过理性设计ORR 电催化剂的尺寸、微观结构、活性位点密度和界面/表面工程,提升催化剂的ORR性能。
在催化剂的稳定性方面,碳基催化剂在质子交换膜燃料电池实际应用装置上存在稳定性差的问题,严重阻碍了材料的商业化进程。稳定性差可能源于碳基底的腐蚀与金属活性中心的脱除、中间体H2O2的氧化反应及微孔水淹等因素。高工作电压下引起的碳腐蚀导致碳基底中CNx位点被破坏,进而引起活性金属位点的溶解、团聚和流失,导致催化剂稳定性降低。H2O2副产物的生成同样造成碳基底的氧化和腐蚀。因此,从工程设计的角度出发,应避免H2O2的生成、减少碳氧化现象;采用多次碳化形成高度石墨化的碳结构、构建石墨层核壳结构[73];引入抗H2O2氧化的金属位点[74]及自由基清除剂[75];使用疏水性更好的膜材料等,利用这些措施来提高催化剂在酸性介质中的稳定性,促进质子交换膜燃料电池的商业化。
此外,碳基催化剂在膜电极中的负载量通常为1~4 mg/cm2,是商业Pt/C催化剂载量(0.2~0.5 mg/cm2)的5~10倍[21],较厚的催化剂层导致低效的传质和电荷传输,产生较大的欧姆极化和浓差极化,降低了其在燃料电池膜电极中的活性和稳定性。因此,需要综合考虑催化剂活性、稳定性和膜电极界面等多种因素,推动碳基非贵金属催化剂在燃料电池领域的大规模商业化应用。
