基于词义增强的生物医学命名实体识别方法
2023-10-17陈梦萱陈艳平扈应黄瑞章秦永彬
陈梦萱,陈艳平,扈应,黄瑞章,秦永彬
(1.贵州大学 公共大数据国家重点实验室,贵阳 550025;2.贵州大学 计算机科学与技术学院,贵阳 550025)
0 概述
随着生物医学文档数量的迅速增长,生物医学信息抽取变得越来越重要。实体作为句子的主体,包含了丰富的语义信息,因此,命名实体识别在生物医学文本的理解和处理过程中具有非常重要的意义。生物医学命名实体识别(Biomedical Named Entity Recognition,BioNER)指识别文本中的基因、蛋白质、化学药品、疾病等生物医学实体。
深度学习技术在自然语言处理(Natural Language Processing,NLP)中的应用促进了生物医学信息抽取的发展。然而,将最先进的NLP 方法直接应用于生物医学信息抽取时效果并不理想,一个重要原因是通用的预训练语言模型主要在包含一般领域文本的语料库上训练和测试,而生物医学语料库和一般领域语料库的词分布有很大差异。因此,越来越多的研究人员通过在生物医学语料库上训练通用预训练语言模型得到生物医学领域特定的预训练语言模型,如BioELMo[1]、KeBioLM[2]、ClinicalBERT[3]、BlueBERT[4]、SciBERT[5]、BioBERT[6]等。这些模型大多采用WordPiece 标记化方法处理未登录词,即将未登录词以频繁出现的语素来表示(如peri-kappa->per、##i、-、ka、##ppa),在很大程度上缓解了未登录词表示信息不足的问题。
BioBERT 是目前在生物医学信息抽取任务中使用最广泛、最有效的预训练语言模型之一,它在生物医学信息抽取的各种任务上表现都超过了BERT 和之前的先进模型。现有的BioNER 模型将从BioBERT 预训练模型获得的语素嵌入表示后,或直接输入神经网络进行训练,或增加额外的特征信息后输入神经网络进行训练,而忽略了在训练和标签预测过程中可能会出现的如下问题:
1)标签不一致问题。一些语素作为独立单词在文本中出现,可能出现词内部标签不一致问题,比如“peri-kappa”,其真实标签为“B-DNA”,在语素级别进行标签预测时,可能会错误地将其部分语素识别为“kappaB”蛋白质实体的语素“ka”、“ppa”,则“perikappa”语素的标签类型既有DNA 类型又有蛋白质类型,最终导致实体识别失败。
2)跨单词标签问题。英文单词被拆分为语素后,词之间的边界变得模糊,从而产生错误依赖关系,将不同单词的语素识别为同一单词的语素,则会导致实体识别错误。如“CD28 signaling cascade…phospholipase A2 and 5-lipoxygenase”在进行表示学习时,蛋白质实体“phospholipase A2”的语素为“p hos ph oli pas e a2”,在标签预测时模型容易将“e a2”识别为疾病实体“EA2”的语素。
3)语义梯度消失问题。将未登录词拆分为语素后,句长变为原来的几倍,在使用神经网络进行句子语义学习时容易出现梯度消失问题。生物医学数据集中存在大量的未登录复合词,会使得这一问题更加突出,通过统计发现,单词被拆分为语素后长句子明显增多,一些句子长度甚至超过了500。
针对以上问题,本文提出一种基于BiLSTMBiaffine 词义增强的方法。首先经过BioBERT 预训练后将单词拆分为语素,得到语素的嵌入表示后同一单词的语素间使用BiLSTM 获取语素的前向和后向序列信息;然后使用Biaffine 注意力机制增强其关联信息并融合为单词表示,将单词表示作为句子级BiLSTM 的输入,使得文本长度得到控制,有效缓解梯度消失问题;最后以单词作为最小单位,使用CRF序列化标注模型获得最终预测标签,规避由语素导致的词内部标签不一致和跨单词标签问题。本文的主要工作有:
1)针对预训练模型对语素进行表示学习导致的标签不一致、跨单词标签和语义梯度消失的问题,本文提出BiLSTM-Biaffine 词义增强方法,该方法对语素表示进行融合学习,能更好地学习句子中的词级语义特征。
2)设计实现基于BiLSTM-Biaffine 词义增强的生物医学命名实体识别模型,通过在BC2GM(BioCreative II Gene Mention)、NCBI-Disease、BC5CDRchem 和JNLPBA 数据集上进行实验,验证BiLSTMBiaffine 词义增强方法的有效性。
1 相关工作
为提升BioNER 的性能,现有相关研究大致分为3 类:1)通过多种神经网络的组合得到新的模型;2)融合多种特征信息;3)通过在生物医学语料库上训练现有预训练语言模型,得到生物医学领域特定的预训练语言模型。
BioNER 任务通常被建模为一个序列标记问题,且已成功地将CRF[7]、CNN[8]、LSTM[9]等模型应用其中。文献[10]在BioNER 中将CRF 作为基础的分类方法;文献[11]提出一种基于深度神经网络结构的BioNER方法;文 献[12]将RNN 模型应用于BioNER,通过使用大型无标记语料库来解决或缓解复杂的手工设计特征问题;文献[13]提出一种使用BiLSTM 学习正字法特征的BioNER 模型;文献[14]开发多任务CNN 模型,并将其应用于现有的各种生物医学命名实体数据集中;文献[15]将BiLSTM-CRF模型用于不相交标签集的BioNER 任务中;文献[16]将n-GRAM 与BiLSTM-CRF 模型相结合,应用于BioNER;文献[17]使用一组BiLSTM-CRF 模型构建联合模型,以便目标模型从其他协作者模型中获取信息,从而减少误报;文献[18]提出一种多任务学习框架,通过重用相应BiLSTM 单元中的参数,来自不同数据集的输入可以有效地共享字符和字级表示;文献[19]提出基于BiLSTM-CNN-CRF 交叉共享结构的BioNER 模型;文献[20]提出DTranNER 模型,将基于CRF 和深度学习的标签转换模型结合到BioNER中。
为了弥补单纯词嵌入包含信息不足的问题,研究人员进行如下探索:文献[21]证实了字符级嵌入的信息对词嵌入信息有很好的补充作用;文献[22]将深度神经网络、CRF、单词嵌入和字符级表征相结合,进行生物医学命名实体识别;文献[23]提出一种基于词嵌入和字符嵌入的LSTM-RNN-CRF 神经网络架构,使用注意力模型将特定领域的预训练单词嵌入和通过字符嵌入查找表函数获得的每个字符嵌入相结合;文献[24]利用CNN 训练单词以获取具有形态特征的字符级向量,将其与从背景语料训练中得到的具有语义特征信息的词向量进行合并。此外,也有研究者通过加入其他词汇信息来提升实体识别的性能,如:文献[25]提出一种改进的基于语言信息嵌入的BiLSTM 网络架构,除了预先训练的单词嵌入和字符嵌入之外,还包含了缩写嵌入和词性嵌入;文献[26]提出基于CNN 和LSTM 组合特征嵌入的BioNER 方法,通过整合从CNN 和BiLSTM 中提取的2 种不同的字符级表示来增强模型性能;文献[27]使用KVMN 将语法信息合并到主干序列标记器中以改进BioNER;文献[28]通过BERT 进行语义提取以获得动态词向量,并加入词性分析、组块分析特征提升模型精度;文献[29]利用CNN 和BiLSTM 提取文本的字符向量,在训练过程中动态计算文本单词的2 种字符向量权重并进行拼接,同时加入词性信息和组块分析作为额外特征。
由于序列模型获取的信息在很大程度上依赖于词嵌入中包含的信息,因此近年来越来越多的研究者将序列模型的关注点转移到预训练语言模型上。常用的通用领域的预训练语言模型Word2Vec[30]、GloVe[31]、ELMo[32]和BERT[33]应用于生物医学文 本时,会产生大量的未登录词,而且生物医学领域的新生词汇也在不断增长,因此,这些模型并不适用于生物医学领域。有研究者在此基础上提出了专门应用于生物医学领域的特定的预训练语言模型,如:文 献[1]利 用PubMed 摘要训练ELMo得到BioELMo;文 献[2]通过在PubMed 摘要上训练BERT 得到KeBioLM;文献[3-4]通过在临床笔记MIMIC 上训练BERT 分别得到了ClinicalBERT 和BlueBERT;文献[5]通过生物医学和计算机科学领域的论文训练BERT 得到了SciBERT;文献[6]通过PubMed 摘要和PMC 文章训练得到了BioBERT。
WordPiece 技术被广泛应用在上述预训练语言模型中,有效缓解了缺少未登录词表示信息的问题,而现有的BioNER 模型将从预训练模型获得的语素嵌入表示后直接输入神经网络进行训练,或增加额外的特征信息后输入神经网络进行训练,在英文命名实体识别上依旧存在训练时无法充分利用子令牌标签信息和WordPiece 破坏单词语义信息的问题,在未登录词占比较高的生物医学领域,这一问题更加突出。为此,本文提出基于BiLSTM-Biaffine 词义增强的生物医学命名实体识别方法。
2 BiLSTM-Biaffine 词义增强模型
图1 所示为基于BiLSTM-Biaffine 词义增强的生物医学命名实体识别模型整体结构。该模型由编码模块、特征提取模块和解码模块3 个部分组成,其中,b、i、j、o和x为张量对应维度的数值。

图1 BiLSTM-Biaffine 词义增强模型结构Fig.1 BiLSTM-Biaffine word meaning enhancement model structure
2.1 编码模块
语素是构成语言的基本单位,也是最小的语法和语义单位。汉语的语素是汉字,而英语的语素是词根词缀。在英语中,不同的单词可能有相同的词根或词缀,更能代表单词的词义,例如IL-2 和IL-2R都是DNA 类型实体,都包含相同的语素“IL”,这有助于识别实体的类别,同时可以使用已知语素表示未知的新生词汇,有利于获取未登录词的信息。在本文中,使用预训练版本的BioBERT 模型来获取文本的语素嵌入表示。
BioBERT 是基于BERT 模型而提出的,是一种用于生物医学文本挖掘的特定领域BERT,在许多生物医学文本挖掘任务中性能表现优于BERT 和以前的模型,其获取词嵌入的过程如图2 所示。
在图2 中:Wi表示句子中的第i个单词;Wij表示第i个单词的第j个语素。文本以单词序列的形式输入BioBERT,然后根据BioBERT 词表将单词拆分为语素,最后以语素为基本单位进行编码获得语素嵌入表示。
2.2 特征提取模块
特征提取模块由单词级的BiLSTM(Word-BiLSTM)和句子级的BiLSTM(Sentence-BiLSTM)构成。单词级的BiLSTM 用于获取单词内部语素的序列信息,句子级的BiLSTM 用于获取句子的上下文信息。LSTM 的计算公式如式(1)~式(6)所示:
其中:σ和tanh()为激活函数;it、ft、ot和ct分别对应t时刻的输入门、遗忘门、输出门和记忆单元;⊗是点积运算;w和b分别代表权重矩阵和偏置向量,它们的下标表示对应的输入门(i)、遗忘门(f)、输出门(o)和记忆单元(c);xt指t时刻输入语素的特征表示;ht表示t时刻的隐藏状态。
2.2.1 BiLSTM-Biaffine
单词被拆分为语素后,容易产生本文概述中描述的问题并且词的内部信息被割裂,可能会丢失部分词义信息,而BiLSTM 能够捕获序列信息以及关联信息,因此,本文模型在同一单词的语素间应用BiLSTM,以获取语素的前向、后向信息,使用Biaffine注意力机制加强语素间的信息交互,最终将语素表示融合为单词表示,计算公式如式(7)~式(9)所示:
其中:fm表示由LSTM 获取的第m个单词语素的前向信息;bm表示由LSTM 获取的第m个单词语素的后向信息;Um和Wm是与输入有关的矩阵;b′m是可选偏置。
2.2.2 Sentence-BiLSTM
为了获取句子级的上下文信息,本文模型还在句子级使用BiLSTM,即Sentence-BiLSTM,将由BiLSTM-Biaffine 得到的单词表示作为Sentence-BiLSTM 的基本单位,能够有效控制句子长度,缓解梯度消失问题。Sentence-BiLSTM 为双向LSTM,前向LSTM 提取每个单词表示的上文信息,后向LSTM获取单词表示的下文信息。Sentence-BiLSTM在t时刻的隐藏状态由前向和后向隐藏状态拼接而成,如式(10)所示:
2.3 解码模块
NER 任务的最后一步是标签解码,它接受上下文相关的表示作为输入并生成与输入序列对应的标签序列。基于CRF 的解码层能够对最终预测的标签进行约束,弥补BiLSTM 无法处理相邻标签之间依赖关系的缺点,以确保得到最合理的标签序列。
将BiLSTM 的输出作为CRF 的输入,以单词为最小单位进行标签预测,规避了在语素级别进行标签预测而产生的词内部标签不一致的问题。给定输入X,输出预测标签的得分,计算公式如式(11)所示:
其中:Ayi,yi+1是矩阵中的元素,表示标签yi转移到标签yi+1的概率;yi表示y中的元素。在输入X确定的情况下,y预测结果的概率计算公式如式(12)所示:
其中:y|X是所有可能的标签组合;y为真实标签。模型最终选择p(y|X)最大的标签序列,这个选择的过程通过对数似然函数实现。训练过程中的似然函数如式(13)所示:
最终从所有概率得分中通过式(14)选出概率最高的标签序列:
其中:Yx表示所有可能的标签序列组合;y*为最终的标签序列。
3 实验验证
3.1 数据集
在BC2GM、NCBI-Disease、BC5CDR-chem和JNLPBA等4 个BioNER 任务中常用的基准数据集上进行实验,验证本文方法在生物医学数据集上的有效性。BC2GM 数据集由生物医学出版物摘要中的20 128 条句子组成,并对蛋白质、基因和相关实体的名称进行了注释。NCBI-Disease 数据集由793 篇PubMed 摘要组成,对疾病提及进行了注释。BC5CDR-chem 是用于BioCreative V 化学疾病关系任务的数据集,对化学实体和提及进行了手动注释。JNLPBA 数据集包含了扩展版本的生物实体识别数据集GENIA 版本3 命名实体语料库中的MEDLINE 摘要,包含了“DNA”“RNA”“Protein”“cell_type”和“cell_line”这5 类实体。各数据集的实体类型及数量统计结果如表1 所示。

表1 数据集统计信息Table 1 Statistics of datasets
数据集采用“B-实体类型”“I-实体类型”和“O”标签进行标记,使用“B-实体类型”标签标注实体的开始,“I-实体类型”标签标注组成实体的中间部分,“O”标签标注与实体无关的信息。
3.2 评估标准
实验采用精确率(P)、召回率(R)和F1 值(F1)3 种测评指标,具体计算公式如式(15)~式(17)所示:
其中:Tp、Fp、Fn分别表示正确识别的实体个数、错误识别的实体个数和识别出的实体总数量。
3.3 实验设置
本文使用预训练版本的BioBERT 模型,所有的神经网络模型都是基于PyTorch 实现的,优化器采用Adam 算法。实验参数设置如表2 所示。

表2 实验参数设置Table 2 Experimental parameters setting
3.4 结果分析
为了验证本文模型的有效性,分别在BC2GM、NCBI-Disease、BC5CDR-chem 和JNLPBA 数据集上进行实验,将本文模型与近年来BioNER 领域具有代表性的序列标注模型进行对比,实验结果如表3 所示,最优结果加粗标注。
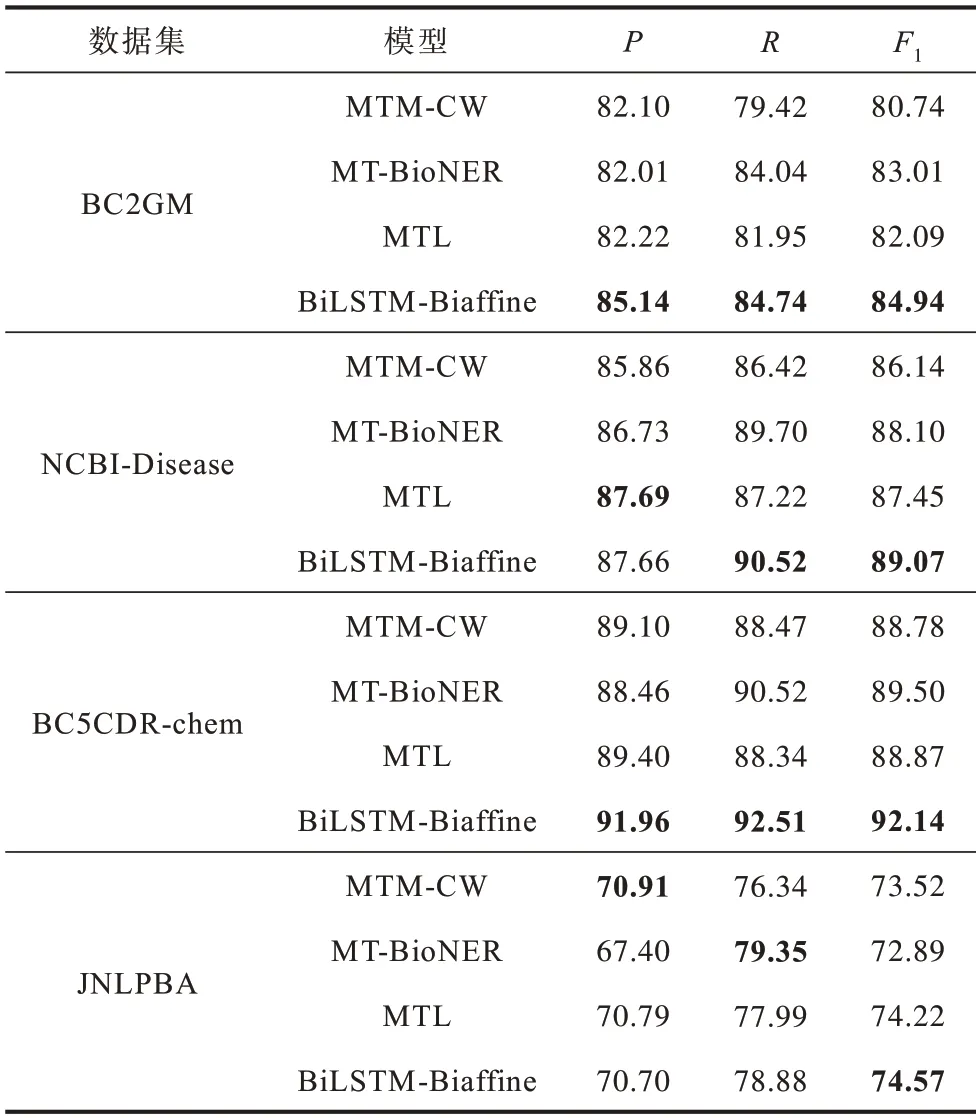
表3 不同模型的对比实验结果Table 3 Comparative experimental results of different models %
在表3 中:MTM-CW 模型是文献[18]提出的融合单词和字符信息的多任务学习模型;文献[34]提出的MT-BioNER 模型结合了BioBERT 和多任务学习;MTL 是文献[35]提出的机器阅读理解模型。以上模型的实验结果均采用了原文献中的最优结果。
从表3 可以看出,BiLSTM-Biaffine 相较于对比模型取得了更优的性能,在BC2GM、NCBI-Disease、BC5CDR-chem 和JNLPBA 数据集上的F1平均分别提升了2.99、1.84、3.09 和1.03 个百分点,并且在数据集BC2GM、NCBI-Disease 和BC5CDR-chem 上的精确率和召回率也都高于对比模型。这主要是因为以下原因:
1)BiLSTM-Biaffine 能够获取BioBERT语素的序列信息以及关联信息。对比模型虽然均使用BioBERT 预训练模型,但是丢失了语素的序列信息和关联信息,BiLSTM 能够获取语素的序列信息,Biaffine 注意力机制加强了其关联并融合为单词表示。
2)BiLSTM-Biaffine 结构能够有效利用丰富的语素信息增强词义表示。字符信息相较于语素包含的信息较少,多任务学习和机器阅读理解无法针对性地增强词义信息,而词义信息对于表示信息相对匮乏的生物医学领域是至关重要的。
3)获取语素的表示信息后将其重新融合为单词表示,不仅解决了未登录词表示信息不足的问题,而且句子长度得到控制,缓解了梯度消失问题。在单词级别进行标签预测,规避了在语素标签预测过程中出现的标签不一致和跨单词标签问题。
为了验证Biaffine 注意力机制的有效性,将其分别与自注意力机制(Self-Attention)、多头注意力机制(Multi-Attention)进行对比,实验结果如表4所示。
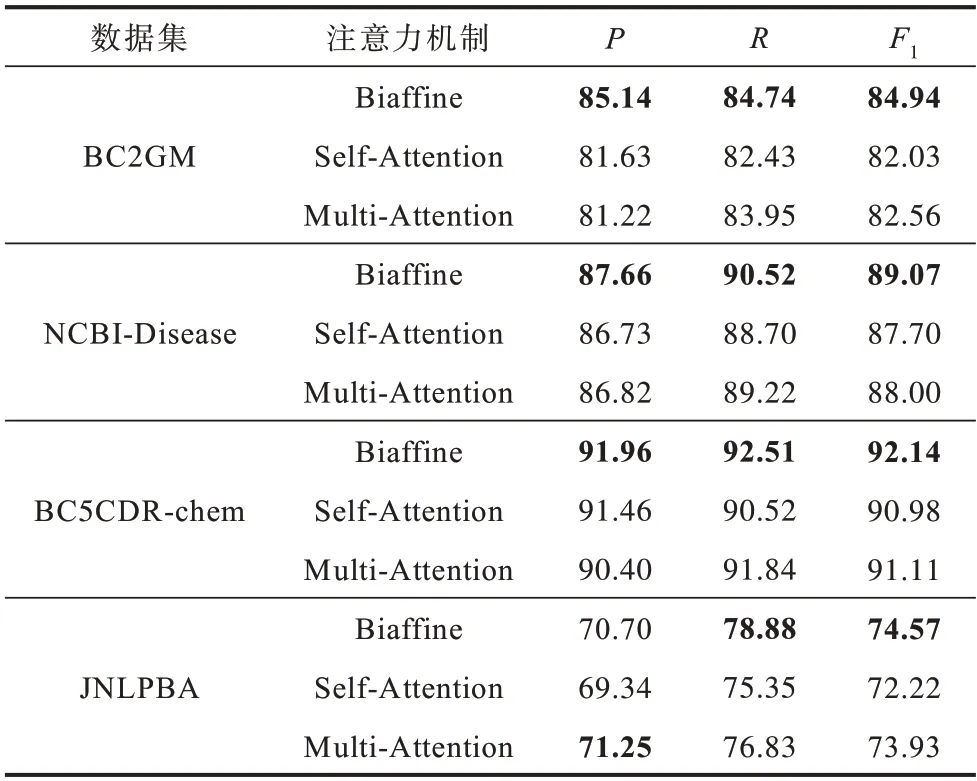
表4 不同注意力机制的对比实验结果Table 4 Comparative experimental results of different attention mechanisms %
从表4 可以看出,相较于Biaffine 注意力机制,Self-Attention 和Multi-Attention 并不能有效提升模型的性能。原因如下:双仿射注意力机制可以进行语素间的信息交互,能够增强同一单词语素间的内部关联信息;自注意力机制和多头注意力机制主要关注怎样从所给输入中捕获重要语义特征,在信息量较少的情况下无法发挥作用。
以上实验结果说明,相较于融合字符信息、多任务学习、机器阅读理解等常用的BioNER 方法,BiLSTM-Biaffine 词义增强方法能够捕获更多的词义信息,使得BioNER 具有更高的准确性。同时,与自注意力和多头注意力机制的对比实验结果也验证了Biaffine 注意力机制的有效性。
3.5 消融实验
为了进一步验证BiLSTM-Biaffine 方法的有效性,本文设计消融实验。在相同的实验环境下,对比BioBERT(模型1)、BioBERT-BiLSTM-CRF(模型2)和BioBERT-BiLSTM-Biaffine-BiLSTM-CRF(模 型3)在数据集BC2GM、NCBI-Disease、BC5CDR-chem 和JNLPBA 上的性能,实验结果如表5 所示。

表5 消融实验结果Table 5 Results of ablation experiment %
从表5 可以看出,在BC2GM、NCBI-Disease、BC5CDR-chem 和JNLPBA 数据集上,模型3的F1相较于模型1 平均分别提高 了1.99、3.31、1.92 和1.36 个百分点,相较于模型2 平均分别提高了0.98、0.99、0.55 和1.36 个百分点。
对模型2 与模型3 达到最优性能所需要的训练批次进行实验,结果如图3 所示。从图3 可以看出,模型3 在4 个数据集上的训练批次均小于模型2,即模型3 具有更快的收敛速度,同时也验证了BiLSTM-Biaffine 结构能够有效缓解梯度消失问题。
相较于BioBERT 命名实体识别模型和BioBERT预训练与BiLSTM-CRF 神经网络模型相结合的命名实体识别模型,本文BioBERT-BiLSTM-Biaffine-BiLSTM-CRF 模型在不使用其他特征信息的情况下取得了更优的性能,并且收敛速度更快,验证了基于BiLSTM-Biaffine 词义增强方法的有效性。
4 结束语
生物医学命名实体识别作为生物医学文本信息的基础任务之一,对于医疗事业的发展有着至关重要的作用。本文从生物医学数据的特殊性出发,提出一种BiLSTM-Biaffine 词义增强方法,以有效利用语素来解决未登录词的表示问题,同时避免由于语素过多导致的文本过长、标签不一致和跨单词标签问题。通过BioBERT 预训练模型获得语素嵌入表示,使用BiLSTM-Biaffine 方法获取语素的序列信息以及完整的单词信息,利用BiLSTM-CRF 模型获取最终的预测标签序列。在BC2GM、NCBI-Disease、BC5CDR-chem 和JNLPBA 等4 个BioNER 任务常用基准数据集上进行实验,结果表明,BiLSTM-Biaffine词义增强方法能够有效提升BioNER 的性能。下一步考虑将BiLSTM-Biaffine 方法应用到其他NER 模型中,并探究一种性能更优的语素融合方法。
