Molecular Stacking and Device Performance:Recent Advances of Efficient Small Molecule Donors Based on Benzodithiophene and Its Derivatives
2023-10-10YANGKeXIAOZeyunLUShirongSUNKuan
YANG Ke, XIAO Zeyun*, LU Shirong, SUN Kuan
Molecular Stacking and Device Performance:Recent Advances of Efficient Small Molecule Donors Based on Benzodithiophene and Its Derivatives
YANGKe1, XIAOZeyun1*, LUShirong1,3*, SUNKuan2*
(,,,400714,;,,,,400044,;,,318000,)
The rapid development of all-small-molecule solar cell has recently gained recognition within the photovoltaic research community. Well-defined structure and less batch-to-batch variation empowered it great application prospects. In this perspective, we review the development of small molecule donors based on benzodithiophene(BDT) and its derivatives, with a focus on the relationship between molecule structure, stacking characteristics and device performance. By analyzing successful cases in the BDT series of small molecules, we aim to clarify the link between molecule structure, solid-state aggregation and device performance. We hope this discussion can be the minnow to catch the whale of highly efficient molecules in the future.
All-small-molecule organic photovoltaics; Benzodithiophene; Molecular stacking
1 Introduction
Developing renewable energy technology is a direct and effective way to address the fossil energy crisis. Among renewable energy sources, photovoltaic power generation is a green and sustainable energy source. With the progress and upgrading, three generations of photovoltaic technologies have been developed. As one of the representatives in the 3rd generation, organic photovoltaics(OPV) has gained attention from the academia and industry due to its unique advantages, such as easy fabrication, flexibility, light weight and tunable band gap. After years of development, the energy conversion efficiency of OPV has exceeded 19% for single junction and over 20% for tandem structure[1,2]. Although the efficiency of OPV has been improved by leaps and bounds in recent years, however, up to now, efficiencies over 18% are all achieved by polymer donor-based OPVs[3—7], which are still suffering from bad batch repeatability and hinder the commercialization of this technology.
Compared with polymers, small molecular materials offer more commercial potential with accurate structure, easy purification and less batch-to-batch variation. Studies on the photovoltaic characteristics of organic small molecules start early[8—12]. But molecular design initially focused on vacuum evaporation deposition, which differs from current needs for high efficiency in solution-processable OPVs. Benzodithiophene(BDT) based small molecules were rapidly developed in last decade to meet these needs. It was first introduced into small molecules by Chen.[13], and have since become the primary choice for high-efficiency small molecule donors. To date, the device efficiency of small molecule based on BDT or its derivatives has exceeded 17% in both binary and ternary structures. Nevertheless, compared to their polymer counterparts, the device efficiency needs to be further improved for all-small-molecule organic solar cells(ASMOSCs) before it can be finally industrialized.
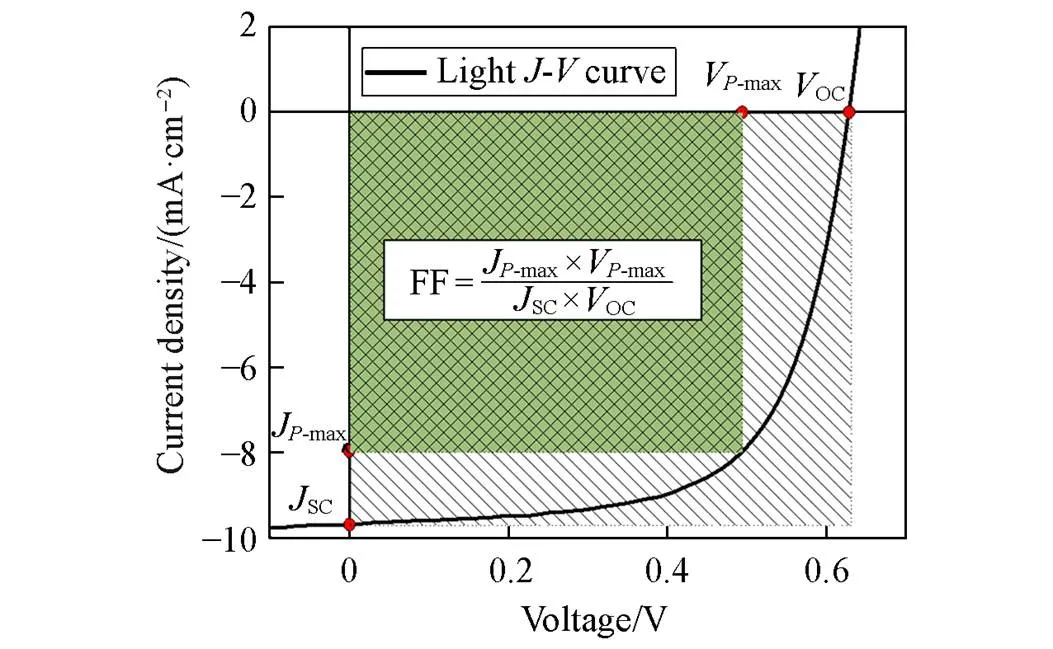
Fig.1 An example of the J⁃V curve of a photovoltaic cell
Academically, the photoelectric conversion efficiency of photovoltaic devices can be described in many ways depending on the operation conditions. In practical applications, photovoltaic modules typically operate under a certain load, so, the ratio of maximum output power to incident light power is often used to represent the energy conversion efficiency of photovoltaic devices. A typical working curve of photovoltaic is shown in Fig.1. Power conversion efficiency(PCE) can be calculated by the following equation.

where theOC(V) is open circuit voltage,SC(mA/cm2) is short circuit current density, FF is fill factor,inrepresent the incident light density, it contains the information of spectrum and intensity. Under different incident light conditions, the device will show different conversion efficiency, with AM1.5G radiation spectrum,inis about 1000 W/m2.V-maxandJ-maxare the corresponding output voltage and current density at the maximum output power(max) point.
Compared with the high-efficiency polymer devices, ASMOSCs usually show lower fill factor and short-circuit current density, which suggests lower carrier yield and charge transfer efficiency. To gain more insight into this disparity, it’s essential to comprehend the charge generation and extraction process in an OPV device. Generally, the working principle of a typical OPV can be separated into 4 steps: light absorption and exciton generation, exciton diffusion to the nearest donor/acceptor interface and the formation of a change tranfer(CT) state exciton, exciton dissociation to generate free charges, charge extraction by corresponding electrode. It’s worth noting that in different perspectives, the steps may be divided into 5 or 6 or more steps, but they are generally included in the above four processes[14,15].
Most reported ASMOSCs have shown lower external quantum efficiency compared to polymer-based devices, but some studies suggest that the primary reason for the low carrier yield is the lower internal quantum efficiency[16]. The difference is mainly stems of the charge separation and transfer process, in conjunction with the lower fill factor that signifies lower charge transfer ability. It can be inferred that the primary limitation is the unsatisfactory phase separation and interpenetrating network structure[1,4,5,17—20]. Due to the smaller conjugated structure of small molecules compared to polymers, charge transport mainly relies on the-interaction between molecules. Therefore, improving the interaction between molecules and regulating the stacking mode and crystallinity of small molecules through material design will be crucial to enhancing device efficiency[21—23].
2 Effects of Molecular Stacking on Device Performance
2.1 Driving Force for Molecular Aggregation
Unlike the crystallization and growth process of inorganic semiconductors, organic molecules are usually assembled by comparatively weak van-der-Waals interactions. The van-der-Waals interaction between two mo-lecules is generated by electrostatic interaction, which originates from the temporal fluctuation of charge distribution caused by the non-perfectly rigid charge distribution in an actual molecule. This temporal fluctuation leads to a fluctuation dipole and generates an attractive force between two molecules. The strength of this force is highly depending on the distance between molecules() and the polarizability() of the molecules, which describes their ability to yield dipole moments. The potential energy of van-der-Waals interaction(vdW) can be estimatedthe following proportional relationships[24].
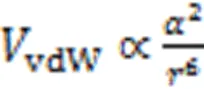
For the needs of solution processing, alkyl or aromatic side chains are usually introduced, which results in greater steric hindrance. According to the van-der-Waals interaction model, the intermolecular interaction is significantly affected. Additionally, different molecular structural design will also produce a variety of steric hindrance and polarizability. As abovementioned, device performance is highly related to the molecular characteristics in isolated state and aggregation properties in solid-state. In the following part, a brief discussion will be carried out with the influence of molecular stacking on charge carrier generation and transport in a typical OPV device.
2.2 Light Absorption
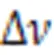

where ℏ andare the Plank constant and speed of light,is the number of aggregated molecules,is the dipole moment of transition in monomer,is the distance between molecule center,is the tilt angle between the line of molecule center and transition dipole moment direction.
2.3 Exciton Dissociation
Due to the low dielectric constant of organic molecules, Frenkel excitons with large binding energy are mainly produced, which cannot be dissociated into free charges under excitation at room temperature. To effectively separate photogenerated excitons, a heterojunction with a certain energy offset is indispensable. It provides sufficient separation energy at the interface by utilizing the energy level difference to release the Coulomb bound. Limited by the short exciton diffusion length in organic materials, bulk heterojunction structure is introduced, which greatly increases the donor acceptor interface thus promote exciton dissociation. At the same time, driven by the different miscibility between donor and acceptor materials, a certain scale of phase separation can form to facilitate charge transfer. Therefore, in order to ensure the effective dissociation of excitons and charge transport, it is necessary to control the phase separation within a certain scale. Generally, the domain size needs to be controlled within several to tens of nanometers respect to the exciton diffusion range of materials[26]. Janssen.[27]proposed that high electron mobility in nanocrystalline PCBM clusters can promote the dissociation of excitons in CT state based on Braun Onsager model. Köhler.[28]proposed that the Coulomb bounding of CT state can be reduced through electrostatic screening and carrier delocalization of polymer donor, so that of CT state can also be dissociated even after thermal relaxation. Friend.[29]proposed thatelectron delocalization of fullerenes may be the key factor affecting the dissociation of CT state.Köhler.[30]proved that C60with higher order and stronger intermolecular interaction has higher electron delocalization and can significantly promote charge separation. It is proved that in addition to the hole delocalization along the conjugated bond, the stacking order and crystallinity of acceptor molecules will significantly affect the intermolecular interaction, and the electron delocalization provided by strong interaction is also very important for charge separation. Although it has been proved that the tight and regular packing of molecules can produce more delocalized charges and promote the dissociation of CT excitons in fullerene systems, in non-fullerene systems, the dissociation of CT state seems to be barrier less, which may be promoted by the quadrupole moments of the acceptors[31,32]. However, at present, conclusive theory is yet widely proved, and the relevant research is still ongoing.
2.4 Charge Transport
When the exciton in the CT state is dissociated into free charges,the charge carrier is transferred through the conjugated backbone or hops to the adjacent molecules through overlapped orbits[33]. Thus this type of charge transfer is heavily affected by molecular packing, including intermolecular distance, aggregation mode, relative orientation, molecular planarity, etc[34,35]. Charge transfer rate(transfer) between two molecules can be described with Marcus theory[36].
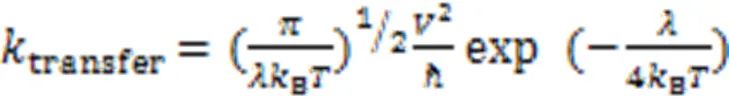

Larger planar conjugate, ordered and closer molecular stacking may lead to better carrier delocalization and more orbital overlap between adjacent molecule, resulting in higher electron coupling and lower reorganization energy, reducing the obstruction of charge hopping and realizing efficient charge transport[38].
3 Embodiment of Molecular Stacking Difference
3.1 Amorphous/Crystalline Aggregation State
The aggregation form in a solid state can be classified as either crystalline or amorphous according to the order of molecular stacking. For example, due to the isotropic characteristics of carbon cages, fullerene and its derivatives usually exhibit amorphous aggregation state, while the materials with planar conjugate structure, including oligomer and polymers, usually intend to aggregate in a specific direction and show the characteristics of anisotropy.
3.2 Orientation
According to the stacking direction of the molecular conjugation plane relative to the substrate, the ordered stacking structures can be roughly categorized as face-on, edge-on and end-on. Generally, when molecules stack along theirconjugation plane and side chain direction, it is referred as-stacking and lamellar stacking[39].
As shown in Fig.2, when the-stacking is perpendicular to the substrate surface, the molecular plane appears to lie flat on the substrate, this type of stacking is called face-on stacking, and sometimes it is also re-cognized by lying down. When-stacking is parallel to the substrate and lamellar stacking is perpendicular to the substrate, the molecules appear to stand on the substrate with one edge, so it is called edge-on stacking. End-on stacking is similar to edge on stacking, where the-stacking is parallel to the substrate, but the long axis of molecules is perpendicular to the substrate, so it is also vividly called standing up stacking. In general, organic molecules are mainly pile up with face-on or edge-on stacking. For BDT small molecules, in most cases, they mainly aggregate with edge-on stacking in neat film. However, when blended with face-on dominant NFA like Y6, most of them will switch to face-on stacking, of course, some will retain the original edge-on stacking to a certain extent to form a mixed orientation. It shows better device performance in occasionally, and it is considered to form 3D charge transfer channel for better performance[40].
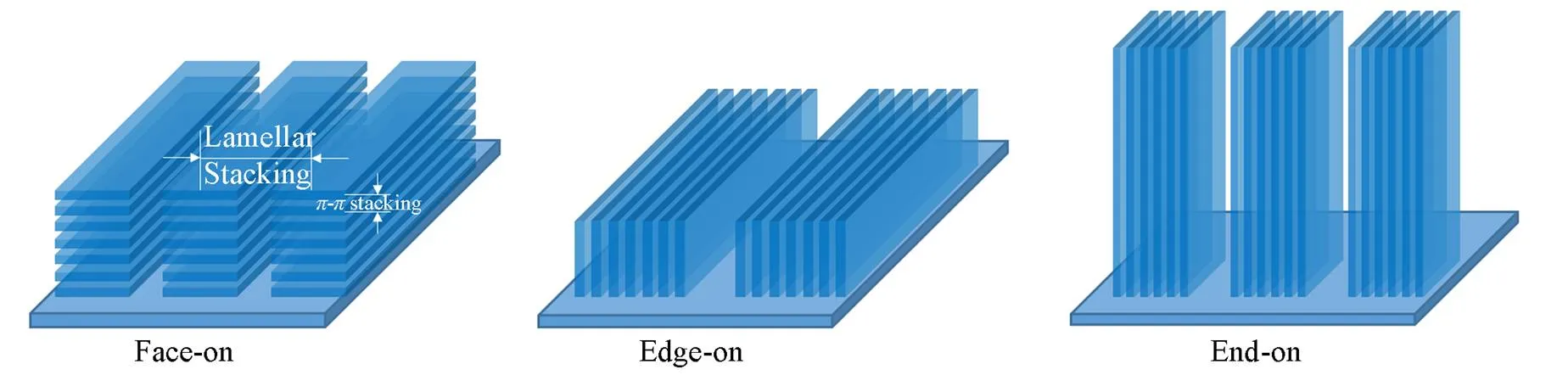
Fig.2 Molecular stacking orientations with respect to substrate
3.3 Aggregation Type
As discussed previously, molecular stacking significantly affects the photoelectric properties of thin films and devices. The stacking mode of two adjacent molecules can be classified into side by side and head-to-tail arrangement, namely H- or J-aggregation[41]as shown in Fig.3. Kwon.[42]summarized the adjacent molecular dipole moments to describe intermolecular interactions. When a molecule is approximated to a point dipole, the potential energy of the interaction between adjacent molecules can be described by a function of the dipole configuration between two molecules as follows.

Fig.3 Excitonic energy state splitting diagram under H⁃ and J⁃aggregation molecule arrangements[41]
Copyright 2011, Wiley⁃VCH.
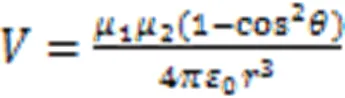
That is to say, two energy levels are split in the excitonic state of this bimolecular aggregate compared with its monomer. They also discovered the significant effect of end-groups on molecular stacking, molecules with asymmetry end-groups tend to form H-aggregation while those with symmetric end-groups exhibit the opposite behavior. The split energy levels in the different aggregation types result in distinct absorption characteristics, many researches have demonstrated a blue-shifted absorption spectrum in H-aggregation and a red-shifted absorption spectrum in J-aggregation. Spano. has modeled and calculated the absorption and emission in different aggregation types, and further clarified the relations between exciton-vibrational coupling and molecular aggregation[43,44].
Based on the understanding of molecular stacking, one can take the advantages of different aggregations,., improve the driving force of exciton dissociation or broaden the absorption spectrum. Cao.[45]have proved the feasibility of broadening the absorption spectrum of the device by enhancing J-aggregation of photoactive molecules. Besides the light harvest, the balance of H-/J-aggregation is also crucial for high performance, by adjusting the vapor pressure and treatment duration in solvent vapour annealing(SVA) process, Han.[41]revealed the importance of H-aggregation and the balance between both packing arrangement in forming well defined morphology and interpenetrating network.
3.4 Packing Size
Toney.[46]summarized the characteristics and quantitative characterization methods of microstructure of organic semiconductor materials at different scales, as shown in Fig.4. The intermolecular interaction takes place in the Angstrom to nanometer scale, which is mainly affected by molecular dipole moment. The aggregation characteristics at this scale will significantly affect the energy level structure of organic semiconductors under condensed state, and then affect the absorption characteristics and carrier delocalization characteristics.

Fig.4 Morphological features and corresponding scales in a bulk heterojunction[45]
The square denotes the enlarged structural diagram under specific size scale.
Copyright 2012, American Chemical Society.
As the scale increases, organic molecules appear in the crystal domain of orderly accumulation in a specific direction. The packing scale is referred to as the crystal coherence length(CCL), which usually ranges from several nanometers. Generally, the CCL is equivalent to the crystallite size and usually estimated by Scherrer equation, which utilizes the corresponding peak width obtained from the grazing incident wide angle X-ray scattering(GIWAXS) test[47,48]. A narrow half-peak width indicates a large crystal coherence length, and. It is noteworthy that, in a stricter sense, the absolute crystal size cannot be directly determined from the peak width of Bragg reflection, as the peak width depends not only on crystal size but also on factors such as crystal size distribution and instrument resolution, especially for those molecules with alkyl chains for better solubilizing, a literally grain size cannot be simply calculated with the peak width of a first- order peak.
Although the method of estimating CCL or crystallite size based on the width of specific diffraction peak is not strictly accurate, it remains an effective and easily applicable and comprehensible method, which also involves less data processing. It is still an effective approach to provide descriptive evidence of relative changes in dealing with different conditions.
To further understand the microstructure of organic semiconductor materials at the scale of tens to hundreds of nanometers, various techniques can be used. For the range of characteristic scale from tens to hundreds of nanometers, GISAXS can usually be used to determine the average domain size of different phases and provide information about domain purity. Similar to the peak width fitting of wide angle scattering, GISAXS cannot effectively provide sufficient contrast for crystalline and amorphous regions due to the small change of mass density in organic semiconductor materials. In other words, the domain size may contain both crystalline and amorphous regions while the CCL is statistical average size of regular crystalline regions.
Since the Frenkel exciton diffusion scale in organic molecules is usually limited by several to tens of nanometers, for the domain size in BHJ, whether the exciton diffusion length can be properly matched determines the carrier separation efficiency in the device. To characterize the microstructure at this scale, in addition to the GISAXS mentioned above, resonant soft X-ray scattering(R-SoXs) and scanning transmission X-ray microscope(STXM) are also effective means to characterize domain size and phase purity.
3.5 Liquid Crystal
Under a certain condition, some organic molecules can form long-range ordered stacking, and maintained the fluidity,., liquid crystalline. Generally, amorphous phase can be considered as a liquid structure fixed on the solid state, while liquid crystal belongs to a fluid with crystal-like long-range order. Usually, the molecules of liquid crystal materials maintain the orientation order through the interaction of electrostatic force and dispersion force. It usually has a main axis with certain stiffness to maintain the shape, and also has some flexible groups or side chains to provide mobility. Due to the strong intermolecular interaction between liquid crystal molecules,well ordered phase is expected, which is conducive to exciton diffusion and charge transfer. These characteristics make the design and application of liquid crystal molecules get extensive attention, and have been widely reported in dye sensitized solar cell(DSSC) and OPV[49—54]. For a newly synthesized molecule, polarizing microscopy(POM) is an essential direct observation method to investigate its liquid crystal property, together with differential scanning calorimetry(DSC) and X-ray characterizations, one can determine whether a molecule has a liquid crystal phase, the classification of liquid crystal phase and related structural information.
There are many ways to classify liquid crystals. According to the molecular characteristics concerned, the types of liquid crystals will be classified in different fields, such as the size of molecular weight(polymer/oligomer/monomer), the way of forming liquid crystal phase(lyotropic/thermotropic), the types of liquid crystal molecules(organic/inorganic/ionic) and the geometry of liquid crystal(rod-like/disk-like). Compared with other classification methods, the nomenclature by molecular arrangement mode under liquid crystal phase(nematic/sematic/etc.) is widely used but less intuitive. The nomenclature and classification of liquid crystal phases started in 1922 by Friedel[55]. Due to the limited number of liquid crystal phenomena observed at that time, it can be classified simply and meaningful. Systems with long range orientation order are referred to as nematic liquid crystal phase, systems with layered structure are referred to as sematic liquid crystal phase. Helical nematic phase, usually observed from the cholesterin derivatives, are referred to as cholesteric liquid crystal phase. However, with more liquid crystal phases found over time, different groups marked the newly observed liquid crystal phases by adding suffix letters, which is easy to cause confusion and misunderstanding. In 2001, the International Liquid Crystal Society(ILCs) and the International Union of Pure and Applied Chemistry(IUPAC) summarized and recommend the nomenclature rules of thermotropic liquid crystals[56].
The photovoltaic effect of organic liquid crystal was first reported by Bard.[50]in 1990 with a porphyrin based liquid crystal. In 2001, MacKenzie.[52]reported a self-organized discotic liquid crystal, hexa-peri-hexabenzocoronene, with a perylene dye,. 2% of PCE was achieved under the illumination at 490 nm. Over 4% of PCE was obtained by liquid crystal molecules in 2013, reported by Adachi.[53]with a liquid crystalline diketopyrrolopyrrole derivative DPP-TP6.
In 2015, BTR is reported as the first liquid crystalline BDT-based small molecule by Jones.[57]. With PC71BM as acceptor, highly efficient ASMOSC was realized with a PCEof 9.3%post SVA treatment. Orderly accumulation and phase separation was activated and realized insensitive film thickness, which is suitable for upscaling roll-to-roll fabrication. Ordered stacking between liquid crystal molecules shows high carrier mobility, ultra-high FF and film thickness tolerance, which shows the potential to meet the needs of roll-to-roll fabrication. With the rise of non-fullerene receptors, many researchers have designed different liquid crystal molecules according to the stacking characteristics of non-fullerenes and made great progress, the details would be discussed vide infra.
4 Design Strategies Towards High Performance Photovoltaics
With the deepening understanding of the relationship between material structure and device performance, small molecule donors have developed different material structures and modification methods. Among the different molecular structures, the small molecule donor with BDT as the core unit is the most prominent, which has attracted more attention and comprehensive structural modification research in the past decade and provide a good spade work for future molecular design. There have been many detailed summaries on the structure of these molecules. Here, we do not intend to simply describe these molecules and their development process again but aim to summarize the past experience of successful molecular modification and put forward our perspectives on future molecular design. In the following part, we will review some BDT based doner and relevant molecule design and modifications.
BDT unit was first introduced into photovoltaic polymers in 2008 by Hou.[58], with the following modifications, BDT-based polymers exhibited outperforming properties and received extensive concern[59—62]. As shown in Fig.5. The exploration of BDT units in small molecule donors began with pioneer works of Chen.[13]. Inspired by the excellent performance of BDT in conjugated polymers, Chen. first introduced BDT units into oligothiophene small molecules in 2011 to increase their core conjugation plane and achieved a breakthrough in efficiency, successfully pushing the PCE of ASMOPVs over 5.4%.

Fig.5 Molecular structure of first BDT based small molecule donor DCAO3T(BDT)3T and its oligothiophene counterpart
It can be seen from the absorption spectra that molecule with BDT core unit exhibits the absorption maxima at 478 and 563 nm in solution or film states, while the oligothiophene shows an absorption maximum at 492 and 580 nm in its monomer or condensed states. After converting the wavelength coordinates of the absorption spectra into photon energy, it can be found that BDT based molecule exhibits more significant bathochromic-shift. As the red shift theory described in Section 2.2, the introduction of BDT significantly affects its molecular stacking characteristics, besides, stronger vibronic absorption peak also indicates that there is a stronger intermolecular interaction. As a consequence, better charge extraction property is realized and higher power conversion efficiency is yielded. Subsequently, small molecule donors with BDT unit have attracted extensive attention and massive research.
4.1 Center Unit
The introduction of large-scale rigid planar conjugate structure can improve the electron delocalization and enhance the molecular stacking, and thus produce better photoelectric property like the introduction of BDT core unit. Similar to the development of thiophene oligomers stems from its polymer counterpart[63—65], the oligomerization of BDT has also been practiced by researchers after the proving of high efficiency in BDT-based polymers[66—71]. As shown in Fig.6, by increasing the number of the BDT core unit and enlarging the conjugate plane, it is expected to increase the number of delocalized carriers and the absorption coefficient of monomers. However, an increase in the number of BDTs will affect the molecular aggregation, as presented by Chen.[66]in 2015. They proposed a BDT trimer based small molecule named as DRBDT3, which exhibited an aggregation feature under solution conditions with high absorption coefficient. While the oligothiophene DERHD7T[72]or the mono-BDT core based molecule DR3TBDT[73], which shared the similar terminal groups with DRB, exhibited featureless absorption pattern in solution state. In aggregated state, oligothiophene and BDT monomer based molecules have significantly greater red-shift than the BDT trimer based one. In addition, while the absorption coefficient of the BDT trimer is higher in the solution state, in the solid state it is comparable to that of the oligothiophene and its BDT monomer counterpart, indicating inferior aggregation. In 2016, Lee.[68]compared the number of BDT units from 1 to 3, and similar changes were observed in their absorption spectra. Compared with a single BDT core, the performance of multiple BDT cores is closer to that of polymers. Therefore, ASMOSCs based on this structure are expected to show better performance in as-cast devices. Ge.[71]prepared as-cast AMSOSC device with an efficiency over 10% based on BDT trimer molecule 3BDT-5, outperformed in reported as-cast ASMOSCs.

Fig.6 Molecular structures of small molecules based on BDT or BDT trimer mentioned above
The aromatic ring expansion based on a single BDT core can also bring greater conjugate plane and carrier concentration like the substitution of naphthalene(naphthodithiophene, NDT) to benzene. As shown in Fig.7, the NDT core was first introduced into OPV by Marks.[74]in 2011, it showed an efficiency over 4% with PC61BM. Since then, NDT based core has been concerned in OPVs. In addition to the conventional modification methods such as side chain,bridge and end group, the relative position relationship between naphthalene ring core and thiophene, such as linear and zigzag configuration, has also been studied[75]. Wei.[76,77]carried out some modification and exploration work on small molecule donors based on NDT core. In 2018, with end group regulation based on NDT system, they prepared two small molecule donors(NDT-3T-R4 and NDT-3T-R6) with a nematic liquid crystalline property. A PCE of 10.4% is realized in NDT-3T-R6∶IDIC-4F blend, which is the best performance based on NDT at present[78]. Due to the complexity of NDT synthesis, there are relatively few relevant research reports. In comparison, the BDT expansions with more thiophene like dithieno [2,3-d∶2′,3′-d′]benzo[1,2-b∶4,5-b′]dithiophene(DTBDT) holds relatively simple synthesis and less isomeric forms, in addition, the conjugation plane is larger than that of BDT and NDT, which is expected to have higher carrier concentration and better stacking characteristics. The well performed DTBDT core in polymers gives an optimistic expectation for its application in small molecules[79]. However, in the early reports, the small molecular devices based on DTBDT are struggling, mainly due to the lack of reasonable phase separation morphology, which hinders the exciton separation and charge transport[80—82]. In 2019, Wei.[83]designed and developed a new DTBDT donor, ZR1. The material showed strong aggregation in solid state. In particular, after thermal annealed under 120 ℃, it showed a crystal-like GIWAXS pattern as collected in Fig.8. Finally, a PCE of over 14% is achieved in ZR1∶Y6 based ASMOSC. Based on this, Wei.[84]developed two small molecules based on DTBDT core and the alkylthiolation phenyl side chain strategy,which showed advantages in BDT small molecules. By modulate the alkyl-thiolation side chain from the-(P-PhS) to the-(M-PhS) position, the dihedral angle of the backbone and the aromatic side chain is reduced, thus reducing the steric hindrance of molecular stacking. The larger red-shifted absorption of M-PhS from solution to film suggests a denser molecular aggregation, which is further strengthened after heat treatment. On the GIWAXS spectrum, M-PhS shows a regular molecular arrangement with spotty scattering pattern, as shown in Fig.8. In addition, M-PhS also showed better miscibility with non-fullerene acceptor(NFA) BTP-eC9, which expected conducive to charge separation. Finally, a PCE of 16.2% was achieved. Recently, on the basis of M-PhS, they optimized the alkyl chain length on the terminal group. When the alkyl chain of the terminal group is shortened from hexyl to ethyl(MPhS-6C/MPhS-4C/MPhS-2C), MPhS-2C exhibits a stacking property that is insensitive to thermal annealing, which significantly inhibits the HOMO energy level shift up during thermal annealing. Shorter carbon chains also bring lower steric hindrance, making the ethyl based molecules have tighter molecular stacking. Notably, with the shortening of alkyl side chain length, non-radiative recombination loss is decreased from 0.242 V(MPhS-6C) to 0.192 V(MPhS-2C), realizing a high open circuit voltage of 0.888 V and a high PCE of 17.11%. This is also the highest efficiency in binary ASMOSCs[85].
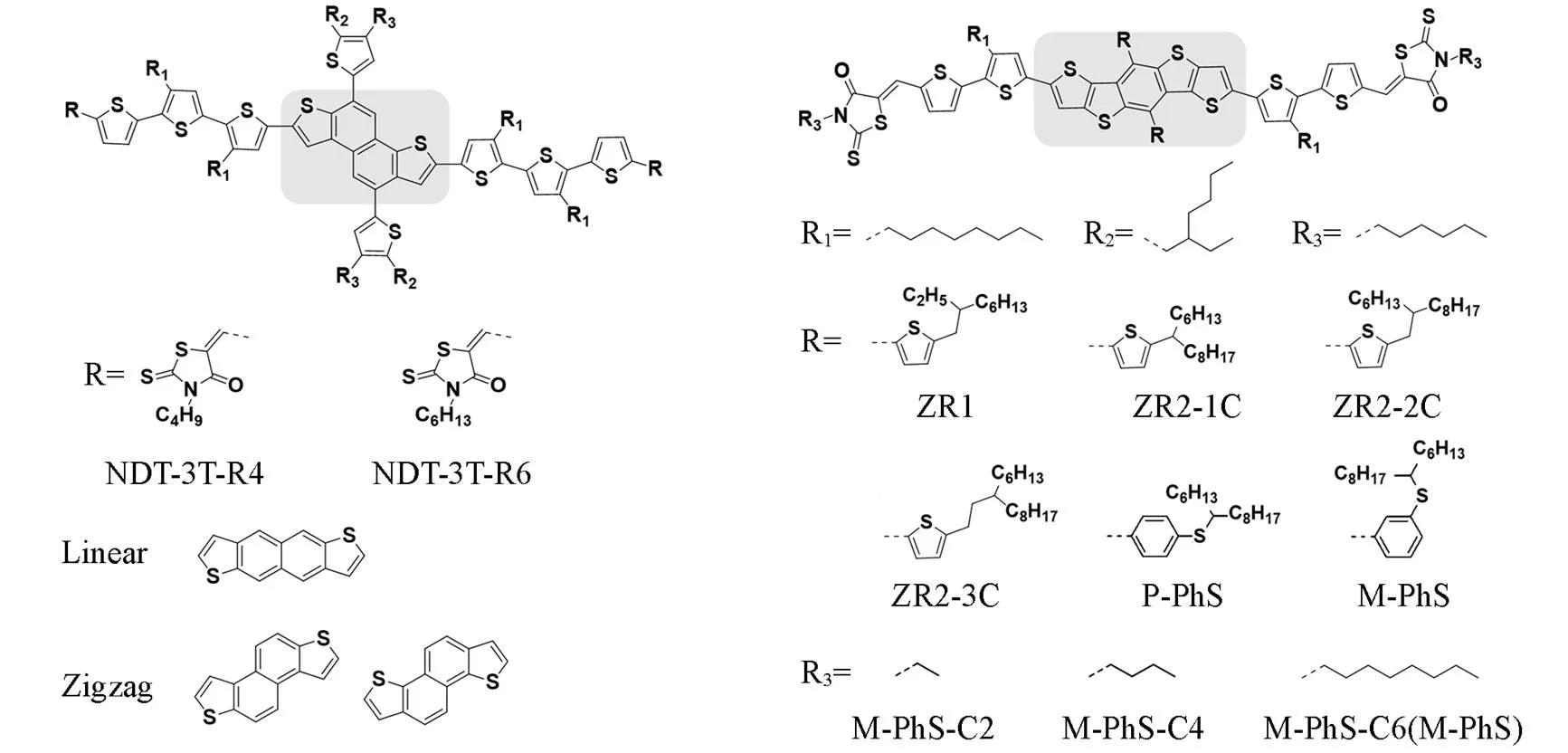
Fig.7 Molecular structures of some high performed small molecule donors with NDT or BTBDT core unit discussed above and the illustration of different NDT isomer structures

Fig.8 GIWAX scattering patterns of as⁃cast and thermal annealed ZR1(A)[83] and thermal annealed P⁃PhS and M⁃PhS neat films(B)[84]
(A) Copyrigh 2019, the authors;(B) Copyrigh 2021, Wiley‐VCH GmbH.
As it has been demonstrated to produce high-performance devices in various studies, the BDT unit still represents the most promising center unit in small molecule donors and the research on the core modification of BDT is still in progress, such as the expansion of its conjugated structure and element substitution. Modifying the core unit alone may not be sufficient to optimize the overall properties. Other parts of the molecule, such as the end groups, the linker units, and the side chain moieties, can also have a significant impact on the molecular properties.
4.2 π Bridge
In addition to the rigid plane of the core unit, the conjugate structure can also be extended by conjugationbridge. It not only provides charge transfer channel as the linker between core unit and end group, but also participates the energy level construction of molecules, influences the planarity of molecules, the aggregation state of molecules, and thus affects the charge transport between molecules.
Based on the star small molecule benzodithiophene-terthiophene-rhodanine(BTR), Jones.[86]designed and synthesized BXR series with different length of thiophenebridges and BTR series with different alkyl chain length. The effects of these variations were investigated from the perspective of materials and devices. Chemical structures of these molecules are illustrated in Fig.9. It can be seen from the absorption spectra that except for BBR and BPR, which formed an H-aggregation dominant characteristics, BXR series molecules show a molecular stacking dominated by J-aggregation. Stronger bathochromic-shift emerged with the increasing of conjugation length and reached the maximum in quaterthiophene(BQR). Comparing the hexyl side chain of BTR, the molecules based on butyl side chain(BT4R) show less J-aggregation characters. When it turns to octyl(BT8R), the molecules are mainly piles up with H-aggregation due to large steric hindrance. The test of POM and DCS can also prove the orderly accumulation of BQR. For BT4R, although only one endothermic peak and one exothermic peak are observed during thermal analysis, the existence of nematic liquid crystal phase can still be observed under POM, which means that there may be a more complex phase transition process that cannot be detected by thermal analysis. Finally, BQR based devices show slightly higher efficiency than BTR, while BT4R based devices show slightly lower efficiency than BTR. It is worth noting that, researchers often judge whether POM is needed for further liquid crystalline investigation by thermal analysis, however, just like this work, some liquid crystalline molecules like BT4R may be missed out.

Fig.9 Molecular structures of π bridge modified BDT based small molecules discussed in this work
Similar to the extension of BDT aromatic structure,bridges with larger conjugated planes are also considered to improve molecular stacking and enhance backbone hole transfer. In 2016, Wei. synthesized BTID-F series molecules, first introduced 2-(thiophen-2-yl) thieno[3,2-b]thiophene asbridge into BDT small molecule system. Stronger aromaticity unit increase IPd, generate highOC. With PC71BM, 11.3% of PCE is realized in BTID-2F∶PC71BM blend, which is the state-of-the-art PCE in inverted ASMOSCs[87].
In 2019, Lu.[88]design and synthesized two BTR analogues, BIHTR and BOHTR, with thieno[3,2-b]thiophenebridge and modified the position of alkyl side chain. By changing the position of alkyl side chain, the planarity of molecules is improved and the molecular stacking is adjusted. It can be seen from absorption spectra from solution to film state, BIHTR shows a greater red-shift, representing a stronger intermolecular interaction, which may inherit from the decrease of steric resistance, realizing a more compact stacking. The periodically arrangement of petaloid pattern morphology also proved the well-aligned molecular packing. Eventually, with Y6 as acceptor, ASMOSC based on BIHTR yields higher PCE(12.3%) than that of BOHTR(10.3%). As we reported previously, different alkyl chain positions on thebridge can significantly affect the aggregation state of molecules. In 2020, Ge.[89]adjusted the alkyl chain position and length of thiophenebridge based on the difluorinated alkylthiophene-substituted BDT core. They designed and synthesized BTEC-2F and BT-2F. Stronger vibrionic peak can be observed in the absorption spectrum of BT-2F film,representing the formation of more ordered aggregation. PCE over 13.8% was realized in BT-2F∶Y6 blend while the BTEC-2F generated an inferior performance of 13.34%. In 2021, through more detailed device optimization, ASMOSC based on BT-2F∶N3 output a PCE over 15.3%[90].
In addition to the studies mentioned above, diketopyrrolopyrrole(DPP)[91], benzo-[1,2-c∶4,5-c′]dithiophene-4,8-diones(BDD)[92], thiazolo[5,4-d]thiazole(TTz)[93]and other structures have also been reported to connect core units and end groups. However, these units serve as not only a bridge for charge transfer in molecules due to their large conjugation plane like BDD or strong electron absorption ability like DPP and TTz, but also the core unit that constitutes the push pull structure, and it may not be appropriate to simply classify them asbridge. Therefore, in this paper, only some simple thiophene based structures which are used as connecting unit are classified asbridge.
The characteristics of thebridge can greatly influence the planarity, molecular stacking, and charge transport of the overall molecule. For example, increasing the conjugation length of thebridge generally leads to stronger intermolecular interactions, improved molecular stacking, and higher charge transport properties. On the other hand, the position and length of the alkyl side chain on thebridge can significantly affect the aggregation state and planarity of the molecule.
Therefore, when designing a new small molecule donor, one should consider the desired optoelectronic properties and choose abridge that can not only provide the necessary charge transfer channel as a linker between the core unit and end group, but also contribute to the energy level construction of the molecule. The length and position of any side chains on thebridge should also be carefully considered to ensure optimal molecular packing and charge transport properties. It is important to keep in mind that the final choice ofbridge will likely depend on a balance between these different factors.
4.3 End Group
In a BDT based small molecule donor, end group is usually an important electron acceptor group in its A--D--A structure. Therefore, the selection and functional design of the end group play a key role in its material and device performance. In addition to affecting the absorption coefficient of the material, it will also affect the solubility and molecular aggregation, and then affect the absorption and carrier migration under condensed state.
As mentioned previously, the research of BDT based molecules began with the development of oligothiophenes. In the research of end groups, the commonly used end groups were first successfully applied on oligothiophenes and then introduced into BDT system. For oligothiophenes, compared with its polymer counterpart, such as P3HT, due to the lack of sufficient conjugation degree, the characteristics is closer to its monomer in terms of energy level and shows a wide band gap, which determines that it is difficult for thiophene based oligomers to achieve the same optical absorption as their polymers. In order to solve the problem of light harvest, Pfeiffer.[94]introduced dicyanovinyl as the end group of pentathiophene in 2006 and constructed ADA structure to reduce the optical band gap of pentathiophene from 2.5 eV to 1.77 eV, which was then applied into double layered heterojunction devicevapor deposition. In solution processable materials, Chen.[64,65,72,95—97]made a lot of pioneering exploration on end group modification. Among the small molecule donors based on BDT core, the common end groups include different functionalized cyanoacetate(DCAOT), indanedione(DINT), rhodanine(DERHDT) and some of their derivatives, as shown in Fig.10. Chen.[97]compared these three end groups in oligothiophene. In the absorption test in solution and thin film, rhodanine based molecules show the maximum absorption red shift from solution to film state, which implies a better aggregation, they also generate the best device performance. After introducing the BDT core, Chen.[73]compared the performance of ethyl rhodanine(DR3TBDT) and octylcyanate(DCAO3TBDT) end groups. The results also proved that ethyl rhodanine based molecules showed a more significant red shift from solution absorption to film absorption. With PCBM as acceptor, DR3TBT carried out a PCE over 7%, while the PCE of DCAO3TBDT based device was less than 5%. Since that, ethyl rhodanine and its derivatives became the dominant end group choices.
With the development of new donor molecules, especially when OPV enters the non-fullerene era, new requirements are put forward for the functionalization and selection of end groups under the new material system.
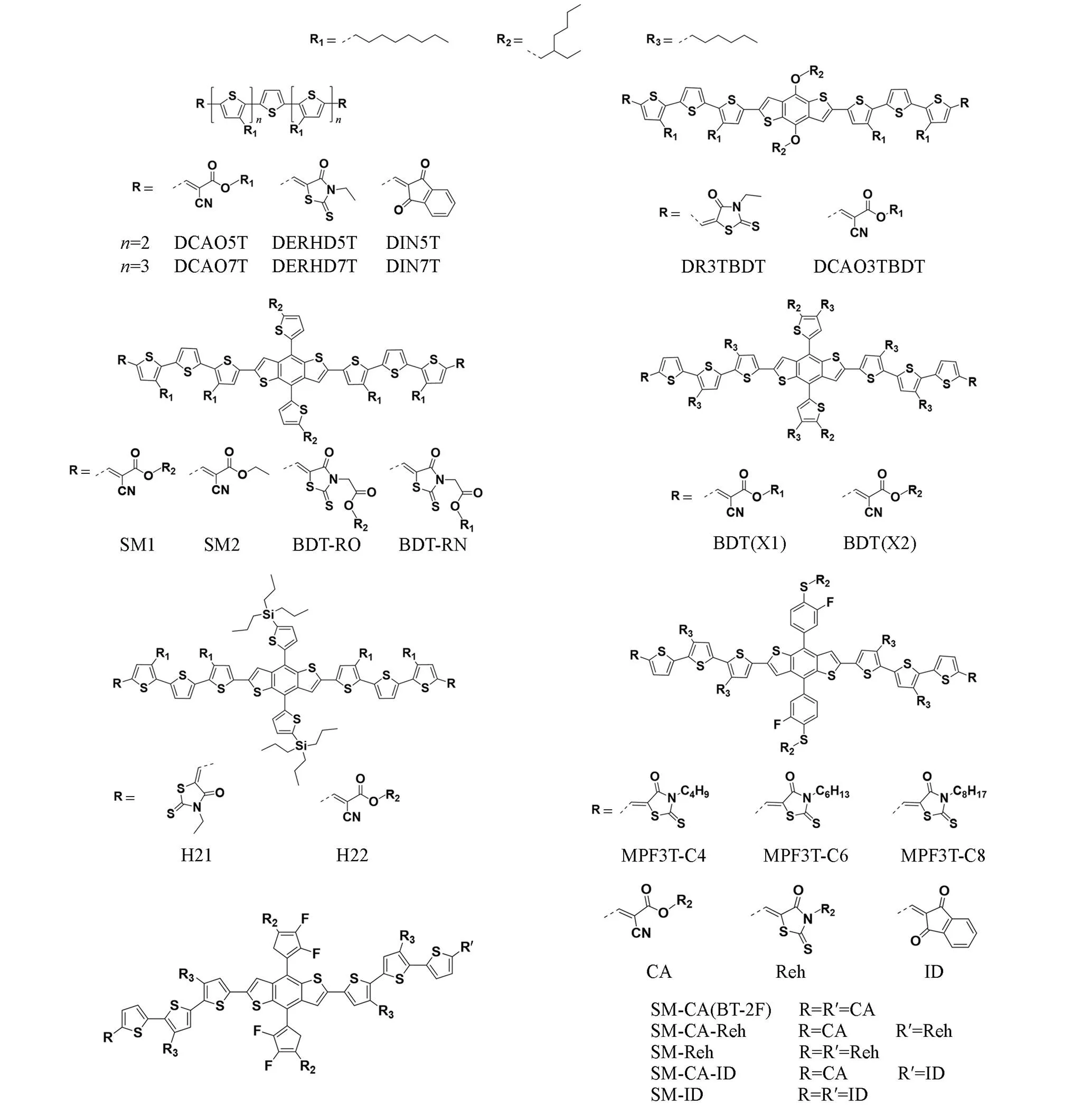
Fig.10 Molecular structures of end⁃group modified BDT based small molecules in this work
In 2017, Li.[98]designed and synthesized two new molecules, SM1/SM2 represent the molecules with/without cyano on their end group, respectively. The effectiveness of ester end group based on cyano free has been proved in small molecular devices. In 2018, they[99]compared the end group performance of ethyl rhodanine(H21) and branched cyano ester(H22) in a BDT based molecule with an alkylsily-thienyl conjugated side chain. The results showed that in this system, although rhodanine based molecules still generate greater red-shift and wider absorption in its neat film, when it comes to the blend film, H22∶IDIC blend showed a wider absorption range. While in H21∶IDIC blend, the absorption range of IDIC showed a certain degree of blue shift and the intensity decreased, which implies that H21 based on ethyl rhodanine end group will significantly affect the molecular stacking of IDIC, and then affect its spectral utilization and charge transport.
In 2019, Lu.[100]designed and synthesized two kinds of BDT based small molecules with esterified rhodanine end groups, namely BDT-RO with 2-ethylhexyl branched alkyl chain and BDT-RN with-octyl straight alkyl chain structure. When blend with NFA acceptor IDIC, branched alkyl chain based BDT-RN shows stronger molecular aggregation and greater phase separation in the blend film, which is difficult to form a continuous interpenetrating network, affecting exciton separation and charge transport. Eventually, BDT- RO∶IDIC yield a better device performance than its branched counterpart.
Since the fullerene and non-fullerene acceptors have different stacking nature, to figure out what kind of end group can match with fullerene acceptor or non-fullerene acceptor. Lu.[101]synthesized two BDT based small molecule donors, BDT(X1) with straight alkyl chain and BDT(X2) with branched alkyl chain[101]. BDT(X1) shows higher crystallinity and suitable for IDIC while the branched X2 is more suitable for PCBM.
In 2021, Hou.[102]carried out fluorine substitution based on their previous reported high-efficiency small molecule donor B1, and regulated the rhodanine end group by adjusting its alkyl chain length from 4 to 8(BPF3T-C4/6/8). Eventually, hexyl showed the best device performance and achieved over 15% of PCE with NFA L8-BO.
Recently, based on the success of BT-2F, Ge.[103]applied asymmetric substitution of end groups to adjust the dipole moment and regulated the stacking properties of molecules. With the introduction of 2-ethylhexyl rhodanine end group, the charge carrier mobility was increased and the HOMO level downshifted, as a consequence, a high PCE of 16.34% are realizedthe improvement ofOCand FF. However, the introduction of indanone end group has indeed led to the decline of device efficiency, which is mainly due to the poor performance of the end group of Indone in this system caused by the excessive accumulation and poor solubility, making it difficult to form a continuous interpenetrating network structure with appropriate size.
The choice and design of end group for a new molecule should be based on a careful consideration with the specific conditions. On one hand, the choice of end group in a small molecular donor can significantly affect its solubility and propensity to form aggregates, both of which can influence its performance in organic solar cells. On the other hand, the impact of the end group on the optoelectronic properties of the molecule should also be taken into account. The electronic nature of the end group can affect the energy levels of the molecular orbitals, which in turn can influence the charge transport and exciton dissociation properties of the molecule.
4.4 Side Chain
The functionalization of side chains is a widely studied molecular structure cutting method in OPV. According to whether the side chain structure is conjugated or not, it can be roughly divided into conjugated and non-conjugated side chains.
The conjugated side chains mainly include aromatic structures such as thiophene and benzene, which can bring extended combination and planar structure. The introduction of conjugated structure will participate in the construction of molecular energy level, affect the molecular energy level structure and carrier transport process to a great extent. Unlike the conjugated side chain, the non-conjugated alkyl side chain is not mainly involved in the energy level construction of an isolated molecule, so the regulation of non-conjugated alkyl side chain has less influence on the photoelectric properties in its monomer state. For instance, in a well dispersed dilute solution, where the molecules are well isolated into monomer, there is no obvious correlation between the absorption characteristics of the molecule and the alkyl side chain. Nevertheless, the alkyl side chain has a great influence on the performance of molecules in aggregated state. The introduction of non-conjugated alkyl side chains was originally to meet the needs of solution fabrication. A certain number of flexible non- conjugated alkyl chains can improve the solubility of molecules, adjust the steric hindrance of molecular stacking, and then change the properties of molecules in the aggregated state. In addition to the aromatic side chain and non-conjugated alkyl side chain mentioned above, some functionalized side chain structures have also been reported to adjust the energy level or-stacking of molecules, mainly including halogenation strategy in non-conjugated structure and homologous element substitution in conjugated structure, such as selenophene or furan substitution and silyl substitution in non-conjugated structure mentioned above.
In 2013, Chen.[104]compared the effects of different conjugated side chain structures and alkyl side chains, synthesized three small molecule donors(DR3TBDTT, DR3TBDTT-HD, DR3TBDT2T) on the basis of DR3BDT. The molecular structures are shown in the Fig.11.
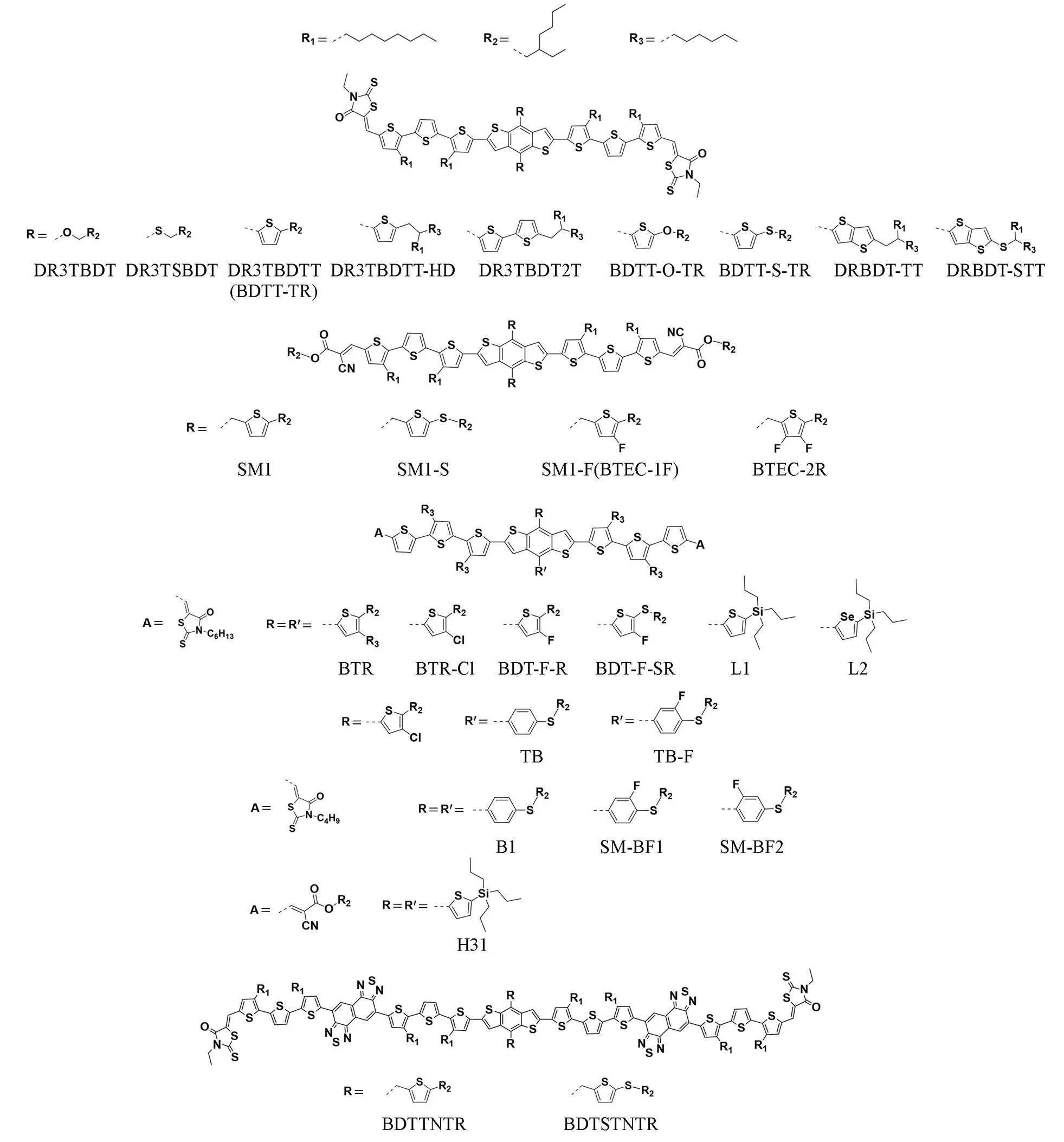
Fig.11 Molecular structures of side chain modified BDT based small molecules discussed in this work
Compared with the alkoxy, larger conjugation plane of the thiophene generates stronger molecular accumulation, but the longer alkyl chain on thiophene will bring greater steric hindrance, making the molecular stacking become H-aggregation dominant. On this basis, further increasing the number of thiophene can alleviate from longer alkyl chain. Based on this, EH monothiophene side chain and HD double thiophene side chain with good stacking performance showed higher PCE up to 8.12% and 8.02%, respectively.
Still based on DR3TBDT, Chen.[111]stdudied the performance of alkylthio(DR3TSBDT) and alkoxy(DR3TBDT) side chains. After the introduction of alkylthio side chain, the HOMO energy level of the molecule is deepened and a higherOCis obtained due to the electron pulling effect of sulfur element. However, the introduction of alkylthio shows a negative impact on the molecular aggregation, the absorption red shift is reduced, and the extinction coefficient is from higher in solution to lower in film state. But after post-treatment, the alkylthio based molecule still yields a high efficiency approaching 10%.
In 2015, Chen.[105]investigated the effects of alkylthio groups(DRBDT-TT) and alkyl groups(DRBDT-STT) in the thiophene side chain with a larger conjugate plane by the introduction of thieno[3,2-b]thiophene(TT) conjugation side chain. Similarly, the molecular stacking based on alkyl side chain is more compact than that based on alkylthio side chain. The-stacking distances are 0.366 and 0.372 nm respectively. The molecules based on alkyl side chain also show higher phase purity in the blend film, generate higher and more balanced carrier mobility, and finally produce higher FF and PCE.
Lu.[106]recently investigated the performance of alkylthio(BDT-F-SR) and alkyl(BDT-F-R) groups in fluorinated thiophene side chian. Similar results are also obtained. After the introduction of thioalkyl side chain, the HOMO energy level of the molecule is deepened and a higherOCis obtained. However, alkylthio shows a negative impact on the molecular stacking. It can be inferred from the GIWAXS test that molecules with alkyl side chains show tighter molecular packing, realizing better charge extraction and power conversion efficiency.
Nevertheless, alkylthio does not necessarily represent a negative impact. In 2016, Min.[107]compared the performance of alkyl(BDTT-TR), alkoxy(BDTT-O-TR) and alkylthio(BDTT-S-TR) on thiophene side chain. In terms of absorption spectra, molecule with alkylthio shows enhanced absorption coefficient, and larger red shift from solution to film, resulting in wider absorption spectrum and short-circuit current density compared with modest performed alkyl side chain. Molecules based on alkylthio side chain also show higher and more balanced carrier mobility. Combined with the characterization results of morphology, alkoxy side chains showed the opposite trend,.., lower absorption and more irregular stacking characteristics. This work shows that under different conjugation systems, the regulation of the side chain may need different substitution strategies.
In 2017, Peng.[108]reported the effectiveness of alkylthio in a BDT based small molecule with naphtho [1,2-c∶5,6 C] bis [1,2,5] thiadiazole as connecting unit between BDT and rhadanine terminal group(BDTTNTR and BDTSTNTR). In terms of energy level and absorption spectra, the effect of alkylthio is similar to that reported by Min.[107]The molecules based on alkylthio side chain show better stacking characteristics and absorption properties. The introduction of sulfur element formed orbital overlap, thus sulfur atoms can accept-electrons, in which divalent sulfur can accept-electrons from the-orbital of adjacent C=C bond into its empty 3orbital[109—111]. The device efficiency based on this molecule exceeds 11.5%, which holds the highest efficiency in fullerene based ASMOSCs.
In addition to the alkylthio side chain strategy, halogenation strategy has also proved to be an effective modification method to improve molecular stacking. In 2019, Lu.[40]reported the chlorination on BTR side chain thiophene(BTR-Cl). With more electron-negative Cl substitution, HOMO energy level is deepened, and the molecules exhibits more ordered molecular stacking property, specifically, high crystallinity and ordered liquid crystalline. As a result, a PCE of 13.6% is achieved with Y6 as acceptor.
Ge.[112]investigated different amounts of fluorination on the thiophene side chain of BDT small molecule(BTEC-1F/2F). More F-substitution deepens the HOMO energy level. However, when the amount of F-substitution exceeded 1, molecular aggregation was weakened with a weak vibrational absorption peak. After heat treatment, absorption profile did not change significantly, but there was a significant red shift, which means that the stacking mode of molecules did not change after heat treatment, but the stacking between molecules became closer. Finally, molecules based on di-fluorinated side chains achieved a PCE of more than 13.3% .
Li’s group[113]investigated different substituents on the thiophene conjugated side chains. They synthesized SM1 for alkyl substituent, SM1-S for alkylthio and SM1-F for fluorine and alkyl substituents. The three molecules exhibited comparable stacking and absorption characteristics. But the electron negativity of sulfur and fluorine deepened the HOMO energy level and improved theOC. Finally, a PCE of more than 14% was achieved based on SM1-F∶Y6 blend.
Wei.[114]studied the effect of alkyl side chain branching position on molecular stacking and device performance with three small molecule donors ZR2-C(=1, 2 or 3), whererepresents the number of linear carbon atoms between branched chain and conjugated moiety. As inferred from the absorption spectra, due to the non-conjugated structure of alkyl chain, which does not participate in the construction oforbital, the change of branching point does not affect the absorption of a single molecule. In dilute solution, all the three molecules show an identity featureless pattern, while the absorption spectrum changes significantly in thin film condition. The molecules based on C1 exhibit H-aggregation, while the other two are mainly J-aggregation. The degree of red shift from solution absorption to film absorption is also different, which means that branching point of alkyl chains has significant impact on molecular aggregation. GIWAX scattering pattern also proved a lower-spacing and larger crystal coherence length in C3, as a consequence, the device shows higher carrier mobility and short-circuit current density and a high efficiency up to 14.37%.
Hou.[115]replaced the asymmetric thiophene unit of BTR with a symmetric phenyl unit, and combined with the alkylthio to regulate the molecular aggregation. The dihedral angle between BDT core and phenyl is larger than that of thiophene and shows a larger rotational potential barrier,., the side chain based on phenyl can significantly affect the molecular aggregation. Dilute solution absorption spectra exhibit that although different conjugated structures are introduced, no significant effect emerges with only lower thiophene absorption characteristics around. 450 nm[25]. In thin film state, B1 shows a similar aggregation mode as BTR, but has greater red shift characteristics, representing the molecules are more closely packed and more conducive to charge hopping between condensed molecules. Compared with BTR, B1 shows higher and more balanced carrier mobility, and finally achieves 15.3% high efficiency with BO-4Cl as acceptor, once the best device performance among ASMOSCs.
Li’s group[116]studied the fluorine substitution at different positions of the benzene side chain of B1. Compared with-fluorination of SM-BF2,-fluorinated SM-BF1 shows a denser molecular aggregation in the in-plane direction. While after blending with Y6, molecule stacking in the blend film is still prevailing by edge-on orientation, which is rarely reported. Usually, BDT based small molecule prefers to form edge-on packing in their neat film, but after blending with Y6, which is heavily face-on oriented, the whole blend usually changes to face-on or face-on dominant mixed orientation. Albeit SM-BF2 shows a face-on orientation that is considered to be conducive to charge transport in vertical direction, its large steric resistance hinders molecular packing and is negative to charge transport. Finally, BHJ based on SM-BF1∶Y6 achieved a high PCE up to 15.71%.
Yang.[117]proposed a 2D side chain symmetry breaking strategyusing different side chain structures. The phenyl side chain structure of B1 and SM-BF1 were introduced into the main structure of BTR-Cl to creat two new asymmetric small molecules,. phenyl substitution(TB) and fluorinated phenyl substitution(TB-F). The introduction of asymmetric structure has no significant effect on the H-aggregation and edge-on orientation nature of BTR-Cl molecule. However, the asymmetric structure brings increased dipole moments and different electrostatic distribution, which trigger an increased crystallinity, dielectric constants and smaller Urbach energy. At the device level, the tighter stacking improves the charge carrier mobility and extraction efficiency, realizing a high FF over 76%, which is one of the highest FF in ASMOSCs. At the same time, the higher dielectric constant improves the exciton dissociation ability and improves the spectral response. Finally, TB-F∶L8-BO based device yielded a PCE over 17%.
In addition to the alkyl chain, tripropylsilyl has been proved to sown-shift the HOMO level in polymers[118]. In 2018, Li’s group[99]introduced tripropylsilyl into BDT small molecule donor and synthesized H21 and H22. Through the end group modification which we have discussed above, efficiency over 10% is obtained with IDIC. In 2021, Janssen.[119]replaced thebridge of H22 with abridge structure similar to BTR, synthesized a new small molecule donor H31, and achieved an efficiency of more than 13.6% with Y6 as acceptor, which proved the applicability of the donor based on tripropylsilyl substitution in Y6 system.
Due to the larger covalent radius, the conjugated structure based on selenium can increase quinoidal resonance, and then reduce the band gap of the material. Secondly, larger and looser outermost electron cloud of selenium makes it more polarizable than sulfur, furthermore, the intermolecular interaction can be enhanced by selenium substitution, thus improving the carrier mobility in the aggregation state. Selenophene substitution has been used in polymers, however, it is yet reported in high efficiency BDT small molecule donors[120—122]. Based on this point, in 2021, Lu.[16]synthesized two new small molecule donors on the basis of liquid crystal molecule BTR and tripropylsilyl side chain, L1 with thiophene side chain and L2 with selenophene side chain. The characterization of POM shows that L1 has a neutral phase similar to BTR, while L2 based on selenophene shows a sematic mesomorphic phase. Rod like crystals can be observed under AFM and STEM test, which means that the introduction of selenophene brings stronger molecular aggregation and carrier mobility. Finally, L2∶Y6 realizes a high efficiency up to 15.8% and still maintains an efficiency over 14% in a thick film with a film thickness up to 300 nm.
The design of side chain is crucial for the photovoltaic performance, larger conjugated side chain may increase the electron density of the molecule, which can bring the possibility of molecular orbital overlap, beneficial to form charge transfer channel. Nevertheless, larger conjugated plane may also affect the solubility, non-conjugated flexible chains need to be introduced to improve the solution processing performance, but the introduction of these non-conjugated structures will also generate problems of steric hindrance, which will affect the molecular aggregation, and impact the device performance. Overall, the choice of side chain can have a significant impact on the performance of OPVs, and careful consideration of the molecular structure and substitution strategies is necessary for optimizing their performance.
5 Summary and Future Perspectives
Although the device efficiency of ASMOSCs has surpassed 17%, additional improvements are required in terms of efficiency, stability, and synthesis simplification before they can be effectively utilized. In terms of efficiency improvement and stability, it can be found from the well performed ASMOSCs that the efficiency depends heavily on their intermolecular interactions.
Close and regular stacking can provide more delocalized charges, which will be conducive to the charge separation and extraction. Tight and orderly stacking often requires molecules to have less steric resistance and lowerplane torsion angle. Therefore, subtle modifications in the molecular side chain will raise a significant difference in the charge distribution and aggregation state and the final photoelectric properties. Nevertheless, these molecular side chains modification methodologies may also depend on specific conjugate systems, this also determines that it is difficult to understand the side chain modification strategies required by different molecular systems once and for all. In the future, more research may be needed to match the newly developed efficient acceptors. The current relevant studies show that the surface electrostatic potential between molecules is an important driving force to help the dissociation of CT excitons in non-fullerene systems[123,124]. Therefore, from the perspective of molecular design, finding small molecular donors that can match the surface electrostatic potential distribution of high-efficiency non-fullerene acceptors is also the key to improve photoelectric conversion efficiency.
From the perspective of conjugated structure, the molecular dipole moment is also an important factor affecting molecular stacking. In addition to common conjugated systems, researchers should also try to find more new conjugated systems, such as phenazine[125], thienodipyrole[126]and other aromatic heterocyclic structures[127,128], which may have different surface electrostatic potential distributions, providing new possibilities for material design. In terms of molecular aggregation, it has been proved that the introduction of element substitution with larger atomic radius such as selenium into the conjugated side chain can not only enhance the molecular accumulation, remold the liquid crystal structure of the molecule, but also provide better charge transport ability. Substitution of other positions in the molecular conjugation structure in BDT system is rarely reported, such asbridge or terminal group. In the future, the optimization of element substitution is also expected to improve efficiency.
Combined with the modification of BDT series small molecules on the side chain, halogenated small molecules usually show better device performance, which means that the introduction of electronegative unit into the core conjugation group may be able to adjust the charge distribution and electron delocalization in the molecule, which is similar to the development of Y series acceptors, Researchers can also consider introducing electrophilic unit into fuse BDT donor core unit. In addition to the above development directions based on experimental reports, computer-aided design based on deep learning is also remarkable. It is expected to bring new ideas and practices in molecular design[129—131]. In terms of synthesis simplification, although the efficiency can be improved to a certain extent through complex functional modification, the complex synthesis also hinders the large-scale application of the materials. Therefore, in the future, it may be necessary to return the idea of molecular design to simple structure and the tradeoff between cost and efficiency.
[1] Cui Y., Xu Y., Yao H., Bi P., Hong L., Zhang J., Zu Y., Zhang T., Qin J., Ren J., Chen Z., He C., Hao X., Wei Z., Hou J.,,2021,(41), 2102420
[2] Zheng Z., Wang J., Bi P., Ren J., Wang Y., Yang Y., Liu X., Zhang S., Hou J.,,2021,(1), 171—184
[3] Liu Q. S., Jiang Y. F., Jin K., Qin J. Q.,Xu J. G., Li W. T., Xiong J., Liu J. F., Xiao Z., Sun K., Yang S. F., Zhang X. T., Ding L. M.,,2020,(4), 272—275
[4] Li C., Zhou J., Song J., Xu J., Zhang H., Zhang X., Guo J., Zhu L., Wei D., Han G., Min J., Zhang Y., Xie Z., Yi Y., Yan H., Gao F., Liu F., Sun Y.,,2021,(6), 605—613
[5] Zhang M., Zhu L., Zhou G., Hao T., Qiu C., Zhao Z., Hu Q., Larson B. W., Zhu H., Ma Z., Tang Z., Feng W., Zhang Y., Russell T. P., Liu F.,,2021,(1), 309
[6] Fu J., Chen H., Huang P., Yu Q., Tang H., Chen S., Jung S., Sun K., Yang C., Lu S., Kan Z., Xiao Z., Li G.,,2021,, 105862
[7] Liao Z., Hu D., Tang H., Huang P., Singh R., Chung S., Cho K., Kumar M., Hou L., Chen Q., Yu W., Chen H., Yang K., Kan Z., Liu F., Xiao Z., Li G., Lu S.,,2022,(14), 7878—7887
[8] Kearns D., Calvin M.,,1958,(4), 950—951
[9] Tang C. W., Albrecht A. C.,,1975,(6), 2139—2149
[10] Tang C. W.,,1986,(2), 183—185
[11] Hiramoto M., Fujiwara H., Yokoyama M.,,1991,(10), 1062—1064
[12] Sariciftci N. S., Smilowitz L., Heeger A. J., Wudl F.,,1992,(5087), 1474—1476
[13] Liu Y., Wan X., Wang F., Zhou J., Long G., Tian J., Chen Y.,,2011,(45), 5387—5391
[14] Vandewal K., Benduhn J., Nikolis V. C.,,2018,(3), 538—544
[15] Kirchartz T., Rau U.,,2018,(28), 1703385
[16] Xu T., Lv J., Yang K., He Y., Yang Q., Chen H., Chen Q., Liao Z., Kan Z., Duan T., Sun K., Ouyang J., Lu S.,,2021,(10), 5366—5376
[17] Song J., Zhu L., Li C., Xu J., Wu H., Zhang X., Zhang Y., Tang Z., Liu F., Sun Y.,,2021,(7), 2542—2552
[18] Nian L., Kan Y., Gao K., Zhang M., Li N., Zhou G., Jo S. B., Shi X., Lin F., Rong Q., Liu F., Zhou G., Jen A. K. Y.,,2020,(10), 2223—2236
[19] Hu D., Yang Q., Chen H., Wobben F., Le Corre V. M., Singh R., Liu T., Ma R., Tang H., Koster L. J. A., Duan T., Yan H., Kan Z., Xiao Z., Lu S.,,2020,(7), 2134—2141
[20] Zheng Z., Hu Q., Zhang S. Q., Zhang D. Y., Wang J. Q., Xie S. K., Wang R., Qin Y. P., Li W. N., Hong L., Liang N. N., Liu F., Zhang Y., Wei Z. X., Tang Z. Y., Russell T. P., Hou J. H., Zhou H. Q.,,2018,(34), 1801801
[21] Tang H., Xu T., Yan C., Gao J., Yin H., Lv J., Singh R., Kumar M., Duan T., Kan Z., Lu S., Li G.,,2019,(21), 1901613
[22] Zhou Z., Xu S., Song J., Jin Y., Yue Q., Qian Y., Liu F., Zhang F., Zhu X.,,2018,(11), 952—959
[23] Lee C. J., Mitchell V. D., White J., Jiao X., McNeill C. R., Subbiah J., Jones D. J.,,2019,(11), 6312—6326
[24] Berland K., Cooper V. R., Lee K., Schröder E., Thonhauser T., Hyldgaard P.,Lundqvist B. I.,,2015,(6), 066501
[25] Yassar A., Horowitz G., Valat P., Wintgens V., Hmyene M., Deloffre F., Srivastava P., Lang P., Garnier F.,,1995,(22), 9155—9159
[26] Halls J. J. M., Arias A. C., MacKenzie J. D., Wu W., Inbasekaran M., Woo E. P., Friend R. H.,,2000,(7), 498—502
[27] Veldman D., İpek Ö., Meskers S. C. J., Sweelssen J., Koetse M. M., Veenstra S. C., Kroon J. M., van Bavel S. S., Loos J., Janssen R. A. J.,,2008,(24), 7721—7735
[28] Bässler H., Köhler A.,,2015,(43), 28451—28462
[29] Gélinas S., Rao A., Kumar A., Smith S. L., Chin A. W., Clark J., Poll T. S. V. D., Bazan G. C., Friend R. H.,,2014,(6170), 512—516
[30] Kahle FJ., Saller C., Olthof S., Li C., Lebert J., Weiß S., Herzig E. M., Hüttner S., Meerholz K., Strohriegl P., Köhler A.,,2018,(38), 21792—21802
[31] Perdigón⁃Toro L., Zhang H., Markina A., Yuan J., Hosseini S. M., Wolff C. M., Zuo G., Stolterfoht M., Zou Y., Gao F., Andrienko D., Shoaee S., Neher D.,,2020,(9), 1906763
[32] Karuthedath S., Gorenflot J., Firdaus Y., Chaturvedi N., De Castro C. S. P., Harrison G. T., Khan J. I., Markina A., Balawi A. H., Peña T. A. D., Liu W., Liang R. Z., Sharma A., Paleti S. H. K., Zhang W., Lin Y., Alarousu E., Lopatin S., Anjum D. H., Beaujuge P. M., de Wolf S., McCulloch I., Anthopoulos T. D., Baran D., Andrienko D., Laquai F.,,2021,(3), 378—384
[33] Burke T. M., McGehee M. D.,,2014,(12), 1923—1928
[34] Bässler H., Köhler A.; Metzger R. M. Ed.;, Springer Berlin Heidelberg, Berlin,2012, 1—65
[35] Nan G., Shi Q., Shuai Z., Li Z.,,2011,(20), 9736—9746
[36] Hutchison G. R., Ratner M. A., Marks T. J.,,2005,(48), 16866—16881
[37] Hutchison G. R., Ratner M. A., Marks T. J.,,2005,(7), 2339—2350
[38] Xu C., Cai P., Zhang X., Zhang Z., Xue X., Xiong J., Zhang J.,,2017,, 136—142
[39] Wang L., Guo S., Zhou K., Ma W.,,2020,(10), 4934—4955
[40] Chen H., Hu D., Yang Q., Gao J., Fu J., Yang K., He H., Chen S., Kan Z., Duan T., Yang C., Ouyang J., Xiao Z., Sun K., Lu S.,,2019,(12), 3034—3047
[41] Zhao Q. Q., Yu X. H., Liu J. G., Xie Z. Y., Han Y. C.,,2016,, 6—13
[42] Kim SO., An T. K., Chen J., Kang I., Kang S. H., Chung D. S., Park C. E., Kim YH., Kwon SK.,,2011,(9), 1616—1623
[43] Spano F. C.,,2010,(3), 429—439
[44] Hestand N. J., Spano F. C.,.,2018,(15), 7069—7163
[45] Xiao L. G., Li Z. D., Hu Q., Liu Y. W., Zhong W. K., Mei X. L., Russell T. P., Liu Y., Min Y., Peng X. B., Cao Y.,,2019,(31), 9618—9624
[46] Rivnay J., Mannsfeld S. C. B., Miller C. E., Salleo A., Toney M. F.,,2012,(10), 5488—5519
[47] Mahmood A., Wang JL.,,2020,(10), 2000337
[48] Xiao Y., Lu X.,,2019,, 100030
[49] Högberg D., Soberats B., Yatagai R., Uchida S., Yoshio M., Kloo L., Segawa H., Kato T.,,2016,(18), 6493—6500
[50] Gregg B. A., Fox M. A., Bard A. J.,,1990,(4), 1586—1598
[51] Petritsch K., Friend R. H., Lux A., Rozenberg G., Moratti S. C., Holmes A. B.,,1999,(1), 1776—1777
[52] SchmidtMende L., Fechtenkötter A., Müllen K., Moons E., Friend R. H., MacKenzie J. D.,,2001,(5532), 1119—1122
[53] Shin W., Yasuda T., Watanabe G., Yang Y. S., Adachi C.,,2013,(12), 2549—2556
[54] Kumar M., Kumar S.,,2017,(1), 85—111
[55] Friedel G.,,1922,(18), 273—474
[56] Barón M.,,2001,(5), 845—895
[57] Sun K., Xiao Z. Y., Lu S. R., Zajaczkowski W., Pisula W., Hanssen E., White J. M., Williamson R. M., Subbiah J., Ouyang J. Y., Holmes A. B., Wong W. W., Jones D. J.,,2015,, 6013
[58] Hou J., Park M. H., Zhang S., Yao Y., Chen L. M., Li J. H., Yang Y.,,2008,(16), 6012—6018
[59] Liang Y., Feng D., Wu Y., Tsai S. T., Li G., Ray C., Yu L.,,2009,(22), 7792—7799
[60] Liang Y., Wu Y., Feng D., Tsai S. T., Son H. J., Li G., Yu L.,,2009,(1), 56—57
[61] Liang Y. Y., Xu Z., Xia J. B., Tsai S. T., Wu Y., Li G., Ray C., Yu L. P.,,2010,(20), E135—138
[62] Hou J., Chen HY., Zhang S., Chen R. I., Yang Y., Wu Y., Li G.,,2009,(43), 15586—15587
[63] Liu Y., Zhou J., Wan X., Chen Y.,,2009,(27), 5209—5215
[64] Liu Y., Wan X., Yin B., Zhou J., Long G., Yin S., Chen Y.,,2010,(12), 2464—2468
[65] Liu Y., Wan X., Wang F., Zhou J., Long G., Tian J., You J., Yang Y., Chen Y.,,2011,(5), 771—775
[66] Ni W., Li M., Wan X., Zuo Y., Kan B., Feng H., Zhang Q., Chen Y.,,2015,(2), 339—346
[67] Yang L., Zhang S., He C., Zhang J., Yao H., Yang Y., Zhang Y., Zhao W., Hou J.,,2017,(5), 1958—1966
[68] Badgujar S., Lee G. Y., Park T., Song C. E., Park S., Oh S., Shin W. S., Moon S. J., Lee J. C., Lee S. K.,,2016,(12), 1600228
[69] Yang L., Zhang S., He C., Zhang J., Yang Y., Zhu J., Cui Y., Zhao W., Zhang H., Zhang Y., Wei Z., Hou J.,,2018,(6), 2129—2134
[70] Jia G., Zhang S., Yang L., He C., Fan H., Hou J.,,2019,(1), 76—83
[71] Yang D., Yu K., Xu J., Zhang J., Zhang J., Gao J., Song W., Li D., Chen Z., Ge Z.,,2021,(16), 10427—10436
[72] Li Z., He G. R., Wan X. J., Liu Y. S., Zhou J. Y., Long G. K., Zuo Y., Zhang M. T., Chen Y. S.,,2012,(1), 74—77
[73] Zhou J., Wan X., Liu Y., Zuo Y., Li Z., He G., Long G., Ni W., Li C., Su X., Chen Y.,,2012,(39), 16345—16351
[74] Loser S., Bruns C. J., Miyauchi H., Ortiz R. P., Facchetti A., Stupp S. I., Marks T. J.,,2011,(21), 8142—8145
[75] Zhu XW., Lu K., Li H., Zhou RM., Wei ZX.,,2016,(8), 1271—1276
[76] Zhu X., Xia B., Lu K., Li H.,Zhou R., Zhang J., Zhang Y., Shuai Z., Wei Z.,,2016,(3), 943—950
[77] Li H., Zhao Y., Fang J., Zhu X., Xia B., Lu K., Wang Z., Zhang J., Guo X., Wei Z.,,2018,(11), 1702377
[78] Li H., Wu Q., Zhou R., Shi Y., Yang C., Zhang Y., Zhang J., Zou W., Deng D., Lu K., Wei Z.,,2019,(6), 1803175
[79] Huo L., Liu T., Sun X., Cai Y., Heeger A. J., Sun Y.,,2015,(18), 2938—2944
[80] Cheon Y. R., Kim Y. J., Back J. Y., An T. k., Park C. E., Kim YH.,,2014,(39), 16443—16451
[81] Song H. G., Kim Y. J., Lee J. S., Kim YH., Park C. E., Kwon SK.,,2016,(50), 34353—34359
[82] Feng H., Li M., Ni W., Kan B., Wang Y., Zhang Y., Zhang H., Wan X., Chen Y.,,2017,(4), 552—560
[83] Zhou R., Jiang Z., Yang C., Yu J., Feng J., Adil M. A., Deng D., Zou W., Zhang J., Lu K., Ma W., Gao F., Wei Z.,,2019,(1), 5393
[84] Zhang L., Zhu X., Deng D., Wang Z., Zhang Z.,Li Y., Zhang J., LV K., Liu L., Zhang X., Zhou H., Ade H., Wei Z.,,2021,(5), 2106316
[85] Zhang L., Sun R., Zhang Z., Zhang J., Zhu Q., Ma W., Min J., Wei Z., Deng D.,,2022,(50), e2207020
[86] Geraghty P. B., Lee C., Subbiah J., Wong W. W. H., Banal J. L., Jameel M. A., Smith T. A., Jones D. J.,,2016,, 2298-2314
[87] Deng D., Zhang Y., Zhang J., Wang Z., Zhu L., Fang J., Xia B., Wang Z., Lu K., Ma W., Wei Z.,,2016,, 13740
[88] Dong X. Y., Yang K., Tang H., Hu D. Q., Chen S. S., Zhang J., Kan Z. P., Duan T. N., Hu C., Dai X. X., Xiao Z. Y., Sun K., Lu S. R.,,2019,(1), 1900326
[89] Gao J., Ge J., Peng R., Liu C., Cao L., Zhang D., Fanady B., Hong L., Zhou E., Ge Z.,,2020,(15), 7405—7411
[90] Ge J., Hong L., Song W., Xie L., Zhang J., Chen Z., Yu K., Peng R., Zhang X., Ge Z.,,2021,(22), 2100800
[91] Huang J., Wang X., Zhang X., Niu Z., Lu Z., Jiang B., Sun Y., Zhan C., Yao J.,,2014,(6), 3853—3862
[92] Huo Y., Gong XT., Lau TK., Xiao T., Yan C., Lu X., Lu G., Zhan X., Zhang HL.,,2018,(23), 8661—8668
[93] Wang Y., Wang Y., Zhu L., Liu H., Fang J., Guo X., Liu F., Tang Z., Zhang M., Li Y.,,2020,(5), 1309—1317
[94] Schulze K., Uhrich C., Schüppel R., Leo K., Pfeiffer M., Brier E., Reinold E., Bäuerle P.,,2006,(21), 2872—2875
[95] He G., Li Z., Wan X., Liu Y., Zhou J., Long G., Zhang M., Chen Y.,,2012,(18), 9173—9180
[96] He G., Li Z., Wan X., Zhou J., Long G., Zhang S., Zhang M., Chen Y.,,2013,(5), 1801—1809
[97] Long G., Wan X., Kan B., Liu Y., He G., Li Z., Zhang Y., Zhang Y., Zhang Q., Zhang M., Chen Y.,,2013,(5), 639—646
[98] Qiu B., Xue L., Yang Y., Bin H., Zhang Y., Zhang C., Xiao M., Park K., Morrison W., Zhang ZG., Li Y.,,2017,(17), 7543—7553
[99] Bin H., Yao J., Yang Y., Angunawela I., Sun C., Gao L., Ye L., Qiu B., Xue L., Zhu C., Yang C., Zhang Z. G., Ade H., Li Y.,,2018,(27), 1706361
[100] Duan T. N., Tang H., Liang R. Z., Lv J., Kan Z. P., Singh R., Kumar M., Xiao Z. Y., Lu S. R., Laquai F.,,2019,(6), 2541—2546
[101] Yu Q., Xu J., Fu J., Xu T., Yan X., Chen S., Chen H., Sun K., Kan Z., Lu S., Xiao Z.,,2021,, 109085
[102] Lv Q., An C., Zhang T., Zhang J., Zhang S., Zhou P., He C., Hou J.,,2021,(7), 1200—1207
[103] Ge J., Hong L., Ma H., Ye Q., Chen Y., Xie L., Song W., Li D., Chen Z., Yu K., Zhang J., Wei Z., Huang F., Ge Z.,,2022,(29), 2202752
[104] Zhou J., Zuo Y., Wan X., Long G., Zhang Q., Ni W., Liu Y., Li Z., He G., Li C.,Kan B., Li M., Chen Y.,,2013,(23), 8484—8487
[105] Kan B., Zhang Q., Liu F., Wan X., Wang Y., Ni W., Yang X., Zhang M., Zhang H., Russell T. P., Chen Y.,,2015,(24), 8414—8423
[106] Chen H., Yang K., Huang P., Hu D., Tang H., Lv J., Li G., Lu S.,,2021,(4), 100061
[107] Min J., Cui C., Heumueller T., Fladischer S., Cheng X., Spiecker E., Li Y., Brabec C. J.,,2016,(14), 1600515
[108] Wan J., Xu X., Zhang G., Li Y., Feng K., Peng Q.,,2017,(8), 1739—1745
[109] Cheng YJ., Luo J., Huang S., Zhou X., Shi Z., Kim TD., Bale D. H., Takahashi S., Yick A., Polishak B. M., Jang SH., Dalton L. R., Reid P. J., Steier W. H., Jen A. K. Y.,,2008,(15), 5047—5054
[110] Cui C., Wong WY., Li Y.,,2014,(7), 2276—2284
[111] Kan B., Zhang Q., Li M., Wan X., Ni W., Long G., Wang Y., Yang X., Feng H., Chen Y.,,2014,(44), 15529—15532
[112] Ge J., Xie L., Peng R., Fanady B., Huang J., Song W., Yan T., Zhang W., Ge Z.,,2020,(7), 2808—2815
[113] Qiu B., Chen Z., Qin S., Yao J., Huang W., Meng L., Zhu H., Yang Y. M., Zhang Z. G., Li Y.,,2020,(21), 1908373
[114] Zhou R., Jiang Z., Shi Y., Wu Q., Yang C., Zhang J., Lu K., Wei Z.,,2020,(51), 2005426
[115] Qin J., An C., Zhang J., Ma K., Yang Y., Zhang T., Li S., Xian K., Cui Y., Tang Y., Ma W., Yao H., Zhang S., Xu B., He C., Hou J.,,2020,(7), 1142—1150
[116] Guo J., Qiu B., Yang D., Zhu C., Zhou L., Su C., Jeng US., Xia X., Lu X., Meng L., Zhang Z., Li Y.,,2021,(13), 2110159
[117] Li Z., Wang X., Zheng N., Saparbaev A., Zhang J., Xiao C., Lei S., Zheng X., Zhang M., Li Y., Xiao B., Yang R.,,2022,(10), 4338—4348
[118] Bin H., Gao L., Zhang ZG., Yang Y., Zhang Y., Zhang C., Chen S., Xue L., Yang C., Xiao M., Li Y.,,2016,(1), 13651
[119] Bin H., Wang J., Li J., Wienk M. M., Janssen R. A. J.,,2021,(14), 2008429
[120] Chang WH., Meng L., Dou L., You J., Chen CC., Yang Y., Young E. P., Li G., Yang Y.,,2015,(3), 562—568
[121] Warnan J., El Labban A., Cabanetos C., Hoke E. T., Shukla P. K., Risko C., Brédas JL., McGehee M. D., Beaujuge P. M.,,2014,(7), 2299—2306
[122] Kronemeijer A. J., Gili E., Shahid M., Rivnay J., Salleo A., Heeney M., Sirringhaus H.,,2012,(12), 1558—1565
[123] Yu R., Yao H., Xu Y., Li J., Hong L., Zhang T., Cui Y., Peng Z., Gao M., Ye L., Tan Z. A., Hou J.,,2021,(18), 2010535
[124] Ma L., Yao H., Wang J., Xu Y., Gao M., Zu Y., Cui Y., Zhang S., Ye L., Hou J.,,2021,(29), 15988—15994
[125] Qi X. N., Dang LR., Qu WJ., Zhang YM., Yao H., Lin Q., Wei TB.,,2020,(33), 11308—11339
[126] Nguyen H. Q., Rainbolt E. A., Sista P., Stefan M. C.,,2012,(4), 425—430
[127] Zhou L., Meng L., Zhang J., Zhu C., Qin S., Angunawela I., Wan Y., Ade H., Li Y.,,2021,(8), 2109271
[128] Zhang T., An C., Bi P., Lv Q., Qin J., Hong L., Cui Y., Zhang S., Hou J.,,2021,(35), 2101705
[129] Sun W. B., Zheng Y. J., Yang K., Zhang Q., Shah A. A., Wu Z., Sun Y. Y., Feng L., Chen D. Y., Xiao Z. Y., Lu S. R., Li Y., Sun K.,,2019,(11), eaay4275
[130] Sun W., Zheng Y., Zhang Q., Yang K., Chen H., Cho Y., Fu J., Odunmbaku O., Shah A. A., Xiao Z., Lu S., Chen S., Li M., Qin B., Yang C., Frauenheim T., Sun K.,,2021,(36), 8847—8854
[131] Zhang Q., Zheng Y. J., Sun W., Ou Z., Odunmbaku O., Li M., Chen S., Zhou Y., Li J., Qin B., Sun K.,,2022,(6), 2104742
分子堆叠和器件性能:基于苯并二噻吩及其衍生物的高效小分子给体研究进展
杨可1,肖泽云1,陆仕荣1,3,孙宽2
(1. 中国科学院重庆绿色智能技术研究院薄膜太阳能电池技术研究中心, 重庆 400714; 2. 重庆大学能源与动力工程学院, 教育部低品位能源利用技术重点实验室, 重庆大学⁃新加坡国立大学可再生能源材料与器件联合实验室, 重庆 400044; 3. 台州学院材料学院, 台州 318000)
有机全小分子太阳能电池由于具备结构精确和批次差异小的特点, 有广阔的应用前景, 近年来该领域的发展备受关注. 本文回顾了基于苯并二噻吩(BDT)及其衍生物的小分子给体的发展. 基于分子结构、堆叠特性和器件性能之间的关系, 分析了小分子BDT系列的成功案例, 旨在阐明分子结构、分子聚集和器件性能之间的联系, 为未来高效分子的设计提供参考.
有机全小分子太阳能电池;苯并二噻吩;分子堆积
O649.5
A
10.7503/cjcu20230123
网络首发日期: 2023-05-05.
联系人简介:肖泽云, 男, 博士, 研究员, 主要从事有机太阳能电池方面的研究. E-mail: xiao.z@cigit.ac.cn
陆仕荣, 男, 博士, 研究员, 主要从事有机太阳能电池方面的研究. E-mail: lushirong@cigit.ac.cn
孙宽, 男, 博士, 教授, 主要从事柔性可再生能源材料与器件方面的研究. E-mail: kuan.sun@cqu.edu.cn
2023-03-21
国家重点研发计划项目(批准号: .2022YFB3803300)、国家自然科学基金(批准号: 62074149, 62074022, 22071238)、重庆英才计划创新领军人才项目(批准号: cstc2020jcyj-msxmX0851)、重庆市杰出青年基金(批准号: cstc2020jcyj-jqX0018, cstc2021jcyj-jqX0015)、重庆英才计划青年拔尖人才项目(批准号: CQYC201903008, CQYC2021059206)、中央高校基本科研业务费(批准号: 2020CDJQY-A055)、国家青年千人计划项目(批准号: R52A199Z11)和重庆市自然科学基金(批准号: cstc2021ycjh-bgzxm0093)资助.
Supported by the National Key Research and Development Program of China(No.2022YFB3803300), the National Natural Science Foundation of China(Nos.62074149, 62074022, 22071238), the Chongqing Talents Plan Leading Talents of Science and Technology Innovation Project, China(No.cstc2020jcyjmsxmX0851), the Chongqing Funds for Distinguished Young Scientists, China(Nos.cstc2020jcyj-jqX0018, cstc2021jcyj-jqX0015), the Chongqing Talent Plan Exceptional Young Talents Project, China(Nos.CQYC201903008, CQYC2021059206), the Fundamental Research Funds for the Central Universities, China(No.2020CDJQY-A055), the National Youth Thousand Program Project, China(No.R52A199Z11) and the Natural Science Foundation of Chongqing, China(No. cstc2021ycjh-bgzxm0093).
(Ed.: W, K, M)
