金属配合物在有机太阳电池中的应用进展
2023-10-10王嘉睿于润楠谭占鳌
王嘉睿,于润楠,谭占鳌
金属配合物在有机太阳电池中的应用进展
王嘉睿,于润楠,谭占鳌
(北京化工大学软物质科学与工程高精尖创新中心, 北京 100029)
有机太阳电池因具有质量轻、色彩丰富及可制备柔性大面积器件等优势而备受关注. 开发高性能的活性层给、受体材料及界面层材料是提升有机太阳电池光电性能的关键. 金属配合物兼具金属配位的自组装有序性和有机分子的结构多样性, 且具有较高的三线态激子密度和较长的激子寿命, 是一类重要的光电功能材料. 随着对不同金属配合物光电性质的不断研究, 越来越多的金属配合物光电材料被应用于有机太阳电池中, 并获得了较高的器件光电性能. 本文综述了基于铂、锌、铱、钌、锆等金属的配合物在有机太阳电池活性层、界面层及添加剂中的应用, 并对其结构-性能关系进行了深入分析, 最后对这类材料面临的挑战与机遇进行了展望, 以便为高性能金属配合物材料的设计及其在有机太阳电池中的应用提供参考与启发.
有机太阳电池;金属配合物;活性层材料;界面层材料
近代以来,煤、石油、天然气占据了世界能源需求份额的绝大部分,但随着能源需求的持续增长和温室效应等环境问题的日趋严重,能源结构的转型与可再生能源的开发显得格外重要. 在碳达峰的背景下,各行各业绿色转型是时代的发展趋势,光伏、风电等新能源取代传统能源是“双碳”的必然要求. 有机太阳电池(OSCs)作为第三代太阳电池技术之一,质量轻、柔性高、可低成本制备大面积器件等优点使其具有广阔的应用前景. 随着新型光伏材料的发展和器件结构的优化,单结OSCs器件的能量转换效率(PCE)达到了19.6%[1]. 虽然近10年来OSCs取得了重大突破,但由于存在载流子迁移率低、能量损失大、稳定性差等问题,其光伏性能仍然低于无机太阳电池,这制约了OSCs的商业化进程. 光伏性能的不断突破离不开新型活性层、界面层材料的研发与器件工艺的优化,因此合理利用相关分子的构效关系来设计和合成高效、稳定的光伏材料是有机光伏领域的重要研究方向.
OSCs器件一般由光活性层、两极界面修饰层、透明导电基底和金属电极组成[图1(A)],其核心结构为由给体和受体材料通过溶液加工所制备的光活性层共混膜. 理想的活性层应该具有给体和受体纳米尺度相分离的互穿网络结构,其中给、受体的互补吸收范围决定了电池的光谱吸收宽度,给、受体间的能级差提供了激子解离与载流子迁移所需的动力,这是决定OSCs光伏性能的关键因素之一. 界面修饰层包括阳极界面修饰层与阴极界面修饰层,阳极界面修饰层常常由涂敷在玻璃基底上的聚(3,4-乙烯二氧噻吩)-聚苯乙烯磺酸(PEDOT∶PSS)构成,阴极界面修饰层通常由真空蒸镀的一层低功函金属钙(Ca)或无机化合物氟化锂(LiF),或具有水/醇溶解特性的有机材料PFN-Br制作,界面层不仅可以选择性传输空穴或电子到达器件两极,还具有增强活性层对光子的吸收能力、优化金属电极功函数及提升器件稳定性的作用. 透明导电电极一般为具有高透光性和良好导电能力的氧化铟锡(ITO). 而金属电极一般由真空蒸镀的Al和Ag等金属制成,用于收集阴极界面修饰层传递来的电子. OSCs的工作机制基于有机半导体材料的光生伏特效应,如图1(B)所示,首先太阳光射入透明导电电极,使得光活性层吸收太阳光产生光生激子,激子再扩散到给/受体材料的界面修饰层处解离成空穴和电子,自由的空穴和电子分别沿界面修饰层进行传递,并被阳极和阴极所收集,产生光电流和光电压. 基于对OSC工作机制的深入理解,开发新型高效给、受体以及界面材料,开展活性层形貌调控以及器件结构工程等研究,是有效提升OSC性能的重要途径.

Fig.1 Device structure of OSCs(A) and the working principle of OSCs(B)
在各类光电材料中,有机金属配合物(OMCs)在有机光电器件各功能层中的应用已表现出了巨大的潜力. 了解OMCs在影响光电性能方面的构效关系是实现更高性能的关键. 向有机聚合物或小分子中引入金属配合物具有如下优势:(1)金属氧化还原中心与共轭配体间的-协同作用和中心配位构型可以对材料电荷转移行为实现有效调控;(2)重金属中心的重金属效应可以突破自旋禁阻,实现从最低单重态(S1)到三重激发态(T1)的系间穿越,其中产生的三线态激子具有更长的寿命与扩散距离;(3)基于不同金属离子的空间构型与配位位点可以作为结构模版对分子立体构型精准调控,金属-金属/金属-配体之间的相互作用可以实现具有特殊结构的分子自组装,从而改善材料的机械、光电性能;(4)过渡金属离子可以通过轨道与配体的,或π轨道相互作用来微调最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)之间的间隙,从而改善分子的吸收光谱. 这些特性使得OMCs可以灵活地应用在OSCs器件中的活性层和界面层中. 本文简要综述了OMCs材料在OSCs中的应用进展,以金属元素为区分,分别介绍了其在光活性层、界面层、添加剂等方面的研究历程,并对其构效关系进行简要分析与总结,最后对未来OMCs在OSCs中的发展方向进行了展望.
1 锌基金属配合物材料
1.1 卟啉锌类聚合物给体材料
卟啉是由4个吡咯环通过亚甲基桥互连而成的大环共轭骨架体系,环上有26个不定域的电子. 作为天然光合作用的主要发色团,卟啉分子具有大平面共轭结构,其核心拥有很强的给电子能力,这使得卟啉材料在蓝光(Soret带)和红光(Q带)区域具有非常强的摩尔吸收系数. 当Zn2+与卟啉配位后, Zn2+的轨道介入卟啉环的和*轨道,使得→,→,→*,→*之间的能带发生重叠,所以卟啉分子及其配合物是一类很好的光采集系统. 同时, Zn作为活泼的金属材料具有很低的电离能,能将其电子转移(或部分转移)到卟啉材料的LUMO能级上,形成较为自由的电子,大大增加了卟啉材料中电子转移效率. 得益于其优异的光电性能,含Zn2+的卟啉配合物被广泛应用于各类有机光电器件中.
在对于含卟啉锌的聚合物研究中,由于卟啉的大体积刚性结构,卟啉锌作为主链单元往往会比其作为侧链单元取得更好的光伏性能. Bo等[2]通过Pd催化的Stille偶联和Sonogashira交叉偶联合成方法,合成了两种锌卟啉-alt-二噻吩并[2,3-b∶3,2′-d]噻吩共聚物(ZP1和ZP2). 在ZP1中,二噻吩并噻吩结构可以降低空间位阻,拓展共轭能力并增强光吸收范围,比含有乙炔基的ZP2表现出更好的热稳定性. 由于ZP2中引入了两个乙炔基,使得聚合物主链的π共轭平面得到拓展, ZP2表现出红移的发射和吸收光谱、更强的Q带吸收以及比ZP1更高的空穴迁移率. 以ZP2∶PCBM共混膜为活性层的太阳电池的PCE为0.30%,显著高于基于ZP1的器件(PCE=0.06%). 2010年, Tan等[3]通过将烷氧基苯基为侧链的卟啉锌与寡聚噻和三噻吩连接合成了聚合物ZP3与ZP4. 基于ZP3的OSC器件可分别获得0.18%的PCE,而由于ZP4具有更宽的吸收范围,相应的器件表现出更高的PCE(0.32%). 同一年, Tan等[4]又报道了两种以卟啉锌为核心、齐聚噻吩为4个共轭臂的星形聚合物给体ZP5与ZP6,其中ZP6以三苯胺作为侧链的封端. 齐聚噻吩的引入拓宽了卟啉聚合物的吸收光谱. 器件测试结果表明,基于无封端ZP5的器件表现出更高的PCE(0.61%). 2011年, Moon等[5]将咔唑和芴单元分别与烷氧基苯基修饰的卟啉锌通过Suzuki偶联合成了具有高相对分子质量的聚合物ZP7和ZP8. 测试结果表明,基于ZP8∶PCBM (1∶6,体积比)活性层的器件在理想条件下达到了0.76%的PCE,高于相同条件下以ZP7为给体的器件(PCE=0.62%).
在上述聚合物中,富电子卟啉锌单元与给电子基团结合倾向于形成D-D'型聚合物骨架. 为了拓宽材料的吸收光谱,研究者设计合成了具有给/受体交替结构的D-A型聚合物,显著的推-拉电子效应增强了共轭主链的电子离域,使得聚合物具有更窄的带隙. 2011年, Harvey等[6]报道了一系列以富电子芳环(噻吩和苯)作为桥连接含Pt(Ⅱ)炔单元与卟啉锌单元的聚合物ZP9~ZP11,作者认为通过引入 Pt(Ⅱ)炔单元构建线性共轭体系可以解除自旋禁阻,使活性层产生三线态激子以提高载流子的寿命与扩散距离,并且通过合理的桥搭配增强聚合物在Q带或其它弱吸收带的光捕获能力. 测试结果表明, ZP9~ZP11的光学带隙范围为1.93~2.02 eV,且观察到聚合物在Q带具有典型的强烈→*荧光发射[图2(A)和(B)],基于ZP11∶PCBM的器件取得了PCE为1.04%、开路电压(oc)为0.77 V、短路电流(sc)为3.42 mA/cm2、填充因子(FF)为0.39的最优表现.

Fig.2 Absorption(black), excitation(red) and fluorescence(blue) spectra of ZP11 in 2⁃MeTHF at 298(A) and 77 K[12](B), UV⁃Vis absorption spectra of ZP12 and ZP13 thin films[7](C), XRD patterns of ZP14—ZP16/PC71BM(1∶2, mass ratio) blend films[8](D), UV⁃Vis absorption spectra of copolymers ZP21 in chloroform solution(D) and in thin films(F)[10]
(A, B) Copyright 2011, Amercian Chemical Society;(C) Copyright 2014, Wiley‐VCH Verlag GmbH & Co. KGaA, Weinheim;(D) Copyright 2015, Amercian Chemical Society;(E, F) Copyright 2016, Elsevier B.V.
由于其较窄的光谱带隙以及结构的刚性,基于卟啉基聚合物所制备的器件光伏性能在早期相对较差,如何合理设计聚合物结构以获得更优秀的光伏性能成为了本领域关注的重点问题. 2014年, Hsu等[7]在2D给/受体单元共轭聚合物侧链上引入了卟啉锌单元,设计合成了两种聚合物给体材料ZP12和ZP13. 卟啉锌侧链单元的引入使得聚合物达到了紫外-可见光区的全色吸收[图2(C)],且在卟啉中位点连接的芳香族侧链在调整聚合物构型与改善相分离形貌上起到了一定的作用,其中基于ZP13的器件的sc和FF大幅度提高, PCE达到8.06%,与未引入卟啉锌的原始聚合物给体相比提高了1.26%. 上述分子结构示于图3中.

Fig.3 Molecule structures of ZP1—ZP13
在众多聚合物给体构筑单元中,吡咯并吡咯二酮(DPP)因具有宽而强的吸收光谱、高载流子迁移率、高度共轭的平面以及易化学修饰等优点而成为构筑高效有机光伏材料的理想选择之一,特别是DPP单元和卟啉锌单元共筑的聚合物给体,可以在紫外-可见光-近红外波段实现良好的吸收互补. 2015年, Li等[8]利用喹喔啉单元与卟啉锌的结合单元(QP)合成了一系列由聚噻吩相连的QP-DPP共聚物ZP14-ZP16. 该结构通过融合喹喔啉可以增加聚合物主链的共面性,对共轭平面的二面角起到限制作用,从X射线衍射分析可以看出这些分子的结晶性能有所提高[图2(D)],并导致了强烈的分子内电荷转移效应(ICT),使这类窄带隙聚合物表现出良好的热稳定性、溶解性与宽吸收性. 器件测试表明具有更高DPP含量的ZP16可获得比其它聚合物更高的光伏性能(PCE=5.07%,sc=11.85 mA/cm2),其可能的原因是大位阻的QP结构会使聚合物结构的有序性降低,当QP含量降低时材料会表现出更好的结晶性能,从ZP16更为尖锐的衍射峰可以佐证这一观点,这是高结晶性共混物的标志. Li等[9]在2016年报道了一种以QP为封端的DPP噻吩共聚物ZP18,与没有QP封端的同种聚合物ZP17相比, ZP18表现出了更强的光吸收与更出色的光伏性能,其PCE达到4.45%,较ZP17提高了0.5%. 基于上述研究, Tan等[10]于2017年报道了一系列基于卟啉锌与DPP的共聚物ZP19~ZP22,作者通过调整聚合物分子 中含卟啉锌单体和DPP单体的数量比来改善聚合物对光的吸收,当卟啉锌与DPP的数量比为1∶4时,聚合物ZP21在400~900 nm波段表现出强烈吸收[图2(E)和(F)],并且相应的器件获得了最大sc(6.14 mA/cm2)和最高PCE(2.44%). 2017年, Bucher等[11]合成了具有卟啉锌-DPP结构的聚合物ZP23,并作为给体与PCBM制备成共混膜,随后他们使用热退火(TA)和溶剂退火(SVA)进行器件优化,使得太阳电池的PCE从先前的4.18%增加到6.44%. 上述分子结构示于图4中.
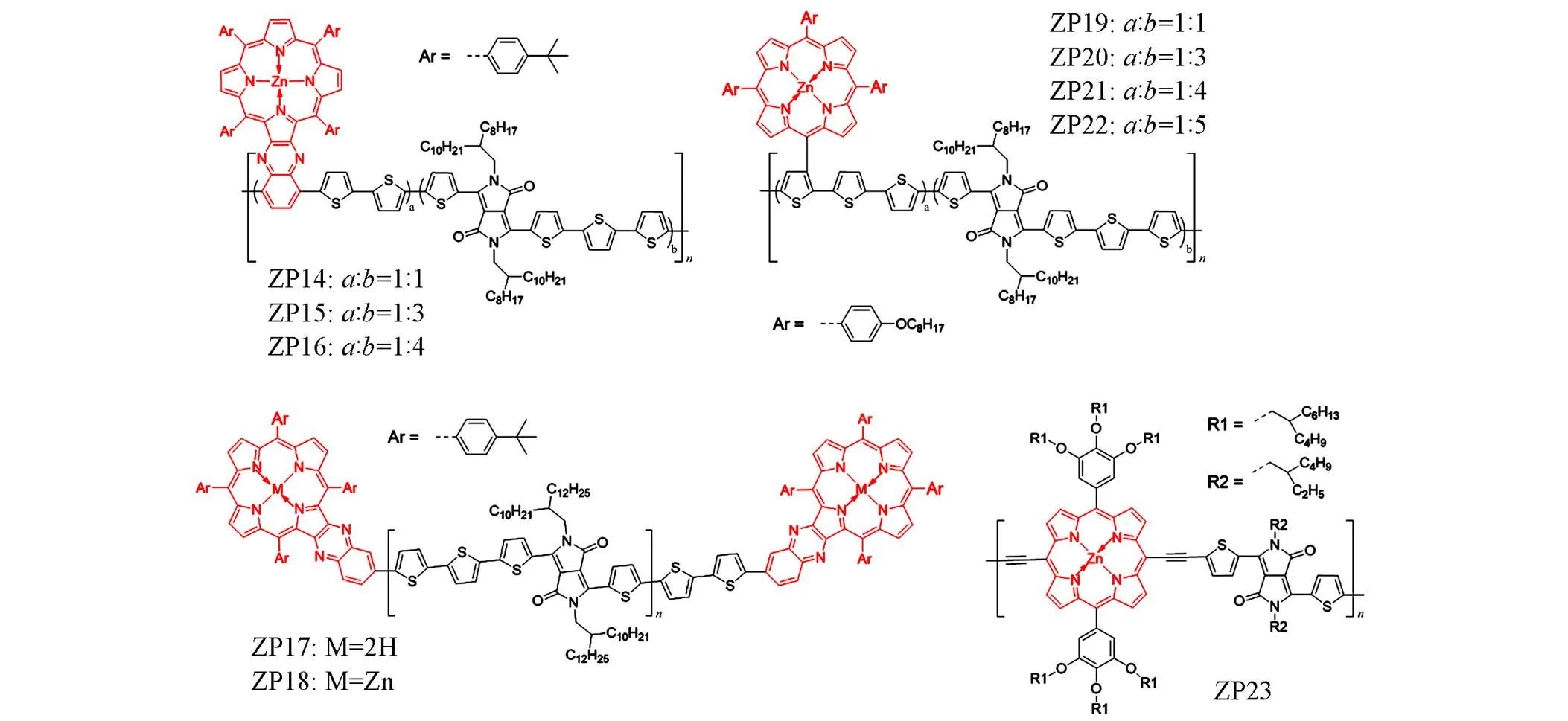
Fig.4 Molecule structures of ZP14—ZP23
1.2 卟啉锌类小分子给体材料
与聚合物材料相比,小分子材料具有合成步骤简单、产物易提纯、分子量分布窄等优势,更重要的是通过对小分子的结构设计可以轻易实现对材料能级、吸收范围、载流子迁移率的调整. 比如,通过给-受体基团的交替构成A-D-A型或A--D--A型排列可以增强分子内电荷转移,调整材料的光学带隙,使器件获得更高的性能,因此对小分子光伏材料的研究也备受关注.
2013年, Coutsolelos等[12]合成了一种含有二苯乙炔结构的卟啉锌小分子ZS1,该分子与PC61BM或PC71BM共混所构筑的太阳电池可获得1.96%或2.54%的PCE. 为了进一步提升基于ZS1∶PC61BM活性层的器件性能,作者采用热退火处理以及使用DMF对PEDOT∶PSS层进行冲洗处理后使器件的PCE最终提高至4.06%. Peng等[13]将卟啉锌与典型的吸电子基团苯并噻二唑(BT)结合,并以3-己基噻吩为封端合成小分子ZS2与ZS3, ZS3分子中卟啉和BT单元之间使用乙炔基作为桥. 在给、受体单元间引入的炔键可以使分子中的卟啉和BT两个单元呈现一个共平面,有利于电子在整个分子中离域并增强分子间的-堆积,最终基于ZS3的器件取得了4.02%的PCE,显著高于基于ZS2的器件(PCE=0.71%). 作为具有强吸电子能力与高平面刚性结构的DPP单元不仅在聚合物材料中广泛应用,在小分子设计中也取得了优异的效果. 2013年, Peng等[14]通过在卟啉锌两侧连接DPP单元并以炔基作为桥合成了小分子ZS4,该结构显著拓宽了材料的吸收光谱至近红外区. 基于ZS4∶PCBM的器件取得3.71%的PCE,此外,通过使用吡啶作为添加剂可以使活性层空穴迁移率得到提高并有效平衡活性层中的载流子迁移,最终实现了4.78%的PCE. 该课题组[15]在此研究的基础上,使用4-辛烷氧基苯基替换了ZS4的 3,5-二(十二烷氧基)苯基合成了小分子ZS5,作者认为ZS4中的烷基链虽然提高了溶解性,但过长的烷基链会抑制分子间的-堆积,降低载流子迁移率. 结果表明,经烷基链修饰后, ZS5的空穴迁移率达到4.68×10‒4cm2·V‒1·s‒1,显著高于ZS4(1.60×10‒4cm2·V‒1·s‒1),基于ZS5的器件取得了7.23%的PCE. 随后,该课题组[16]还研究了在ZS5分子中使用其它硫族原子替换DPP中的硫原子对材料性能及器件光伏性能的影响,合成了基于呋喃和硒的小分子ZS6与ZS7. 得益于重原子的使用降低了带隙并扩展了吸收光谱,基于ZS6或ZS7的太阳电池分别获得了4.3%或5.8%的PCE. 2015年,该课题组[17]研究了烷基链对基于DPP卟啉锌材料光伏性能的影响,并合成了ZS8~ZS10系列小分子. 基于EH侧链的ZS8小分子表现出907 nm的宽吸收范围, 0.78 V的oc以及更低的能量损失. 对基于ZS8∶PC71BM器件进行优化后获得了8.36%的PCE. 此外,与具有更长烷基链的ZS10(PCE=8.24%)相比,基于ZS9∶PC71BM的 器件取得了更高的PCE(9.06%). 作者[18]将基于ZS9∶PC71BM的器件作为子电池制备了叠层电池,如 图5(A)和(B)所示,得益于ZS9分子在400~900 nm的全色光吸收能力与更高的电流密度,该电池获得了12.5%的高PCE,证明了小分子卟啉锌光伏材料的应用潜力. Langa等[19]基于上述材料使用直链烷基作为侧链合成了小分子ZS11,获得了7.04%的PCE,这也进一步阐明了烷基链对调控分子聚集行为的重要作用. 2017年, Peng等[20]基于原有研究报道了一种以苯并噻吩为侧链的小分子ZS12,经过TA与SVA等优化处理后获得了9.08%的高PCE. ZS4~ZS12分子均具有相同的主干结构,可用于研究不同烷基链或芳香族烷基链作为侧链对分子聚集行为的调控,这对分子间电荷运输的研究具有重要意义.
在对卟啉锌衍生物小分子的研究中,研究者对含有推-拉电子单元的小分子结构进行了许多尝试,其中基于四取代的星形结构往往不能取得高PCE,研究者认为4个吸电子基团连接卟啉锌核心后会降低卟啉核心的电子密度,使其光捕获能力变弱,因此研究者认为在卟啉锌的两个相对的中位点连接推拉电子基团来调节共轭与能级,其余两个中位点连接烷基或芳基侧链来调节溶解度或分子堆积是最优的小分子设计理念. 基于此,具有更长共轭结构的A--D--A型小分子成为了领域内的关注热点. 2016年, Langa等[21]使用2-乙炔噻吩作为桥, 3-乙基罗丹宁作为端基合成了两个小分子给体ZS13与ZS14. 作者认为使用强吸电子单元罗丹宁可以增强分子内电荷转移与光吸收能力,其中ZS14由于具有更强的共轭长度显示出了更好的效果. 作者通过原子力显微镜(AFM)对两种材料与PC71BM制备的共混膜进行表征, ZS14的均方根粗糙度为0.53 nm,低于ZS13(0.62 nm), ZS14∶PC71BM共混膜表现出了更好的相分离形貌,基于ZS14∶PC71BM的器件也取得了3.91%的PCE,高于基于ZS13的器件(PCE=3.01%). 为了研究烷基取代基对小分子成膜性以及器件效率的影响, Wong等[22]合成了一系列基于乙炔基苯为桥, 3-乙基罗丹宁为端基,不同烷基侧链长度取代的A--D--A型小分子ZS15~ZS17. ZS15~ZS17的光学带隙在1.50~1.55 eV之间. 如图5(C)和(D)所示,作者发现使用吡啶作为添加剂可以显著降低活性层薄膜的粗糙度,从而提高器件性能,基于ZS15的器件在使用吡啶添加剂后PCE由3.19%提升至6.49%,基于ZS17的器件在使用吡啶添加剂后取得了5.12%的PCE. 而烷基链的结构对于器件性能有直接影响,通过紫外-可见光吸收光谱可以看出,固体ZS16分子与其余两者相比在414 nm处出现了额外的肩峰,这表明侧链方向的改变造成了显著的分子平面差异性. 虽然基于ZS16的共混膜与前两者相比具有较宽的吸收范围,但由于差的结晶性与厚度不均匀等因素仅取得了2.53%的PCE,随后, Peng等[23]基于此研究报道了含有芳基或其它长度烷基取代基的小分子ZS18~ZS20,结果表明卟啉环上不同的取代基显著影响着分子间相互作用,其中基于ZS20的器件取得了最高PCE(7.70%). 为了研究不同吸电子端基对器件性能的影响, 2016年, Langa等[24]设计合成了以噻吩乙烯基噻吩为桥、 3-乙基罗丹宁为端基的ZS21和以二氰基乙烯为端基的ZS22,当使用吡啶作为添加剂进行器件优化后,器件的PCE分别达到5.14%(ZS21)和6.06%(ZS22);当使用Li-TSFI作为添加剂时,基于ZS22∶PC71BM的器件达到了7.63%的PCE. 同年, Peng等[25]使用3,3″-二己基三噻吩作为桥,分别以3-乙基罗丹宁与双氰基罗丹宁作为吸电子端基合成了小分子ZS23与ZS24. 这两种小分子给体在与PC71BM制成共混膜后所制备的器件分别获得了7.66%与8.21%的PCE,可推断使用吸电子效应更强的双氰基罗丹宁作为端基时可以获得更优的光伏性能.
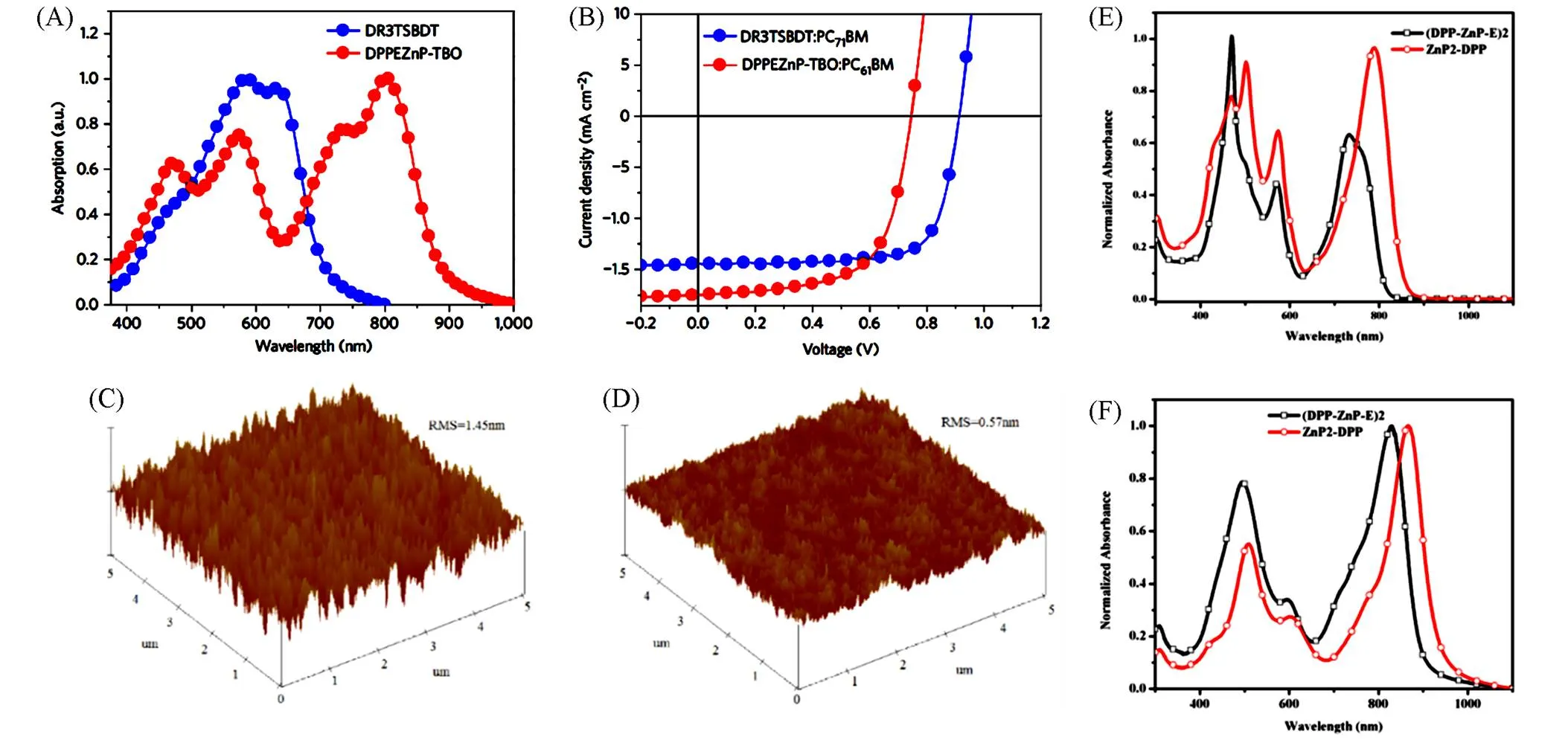
Fig.5 Photovoltaic performance of the single⁃junction devices for ZS9[18](A, B), AFM height images and AFM images of ZS16/PC71BM(1∶1) composite film prepared without and with 3% pyridine[22], respectively(C, D), UV⁃Vis⁃NIR absorption spectra of ZS31 in THF solution(E) and thin film(F)[28]
(A, B) Copyright 2016, Nature Publishing Group;(C, D) Copyright 2015, the Royal Society of Chemistry;(E, F) Copyright 2018, American Chemical Society.
以上介绍的小分子给体均采用了乙炔噻吩基作为桥,对共轭臂上的噻吩基进行共轭长度的改变或取代基的修饰可以促使分子间进行更好的-堆积并增强吸收. 随着研究者对小分子材料设计的不断精进,以环戊联噻吩(CPDTs)作为桥的结构也引起研究者的关注, CPDTs与联噻吩相比具有固定的刚性平面、高导电性与窄带隙,在ICT方面比联噻吩更具优势. 基于此构型, Langa等[26]在2017年设计了以乙炔基CPDTs为桥、分别以3-乙基罗丹宁与丙二腈为端基的小分子给体ZS25和ZS26,基于这两种材料的器件均显示出了低于0.55 eV的能量损耗,其PCE分别为6.71%与7.93%. 特别是将ZS25与具有相同端基的ZS13进行比较时可以发现,使用CPDTs替换噻吩基可以显著提高材料的光伏性能,这归因于具有CPDTs结构的分子往往具有更好的共轭度与平面性,从而获得更高的空穴迁移率,有利于提升器件的sc值.
与单个卟啉相比,由两个卟啉单元连接形成的二元卟啉化合物在分子内能量/电子转移、双光子吸收、非线性光学等方面具有更为独特的性质,因此基于二元卟啉锌的小分子给体被设计并应用于太阳电池中,获得了比单卟啉类材料更高的效率. 2017年, Langa等[19]设计了一种以DPP为吸电子核心的D-A-D型小分子给体ZS27,基于ZS27∶PC71BM的器件获得了8.03%的高PCE,且具有较低能量损失以及平衡的载流子传输性能. Peng等[27]设计合成了3种基于二聚卟啉结构且以DPP为端基的小分子给体ZS28~ZS30,两个卟啉单元分别以二乙炔基、二乙炔基二噻吩以及二乙炔基苯基作为桥接. 将这3种给体分别与PC61BM共混制备太阳电池,分别获得了4.5%(ZS28 )、 5.5%(ZS29)以及6.42%(ZS30)的PCE. 基于上述研究,该课题组[28]又设计了以乙炔基桥接卟啉单元的小分子ZS31,与在四氢呋喃溶液中相比, ZS31薄膜器件显示出了超过1000 nm的近红外吸收[图5(E),(F)]与66%的外量子效率(EQE)响应最高值,sc达到了19.65 mA/cm2,最终获得了8.45%的高PCE,这是当时基于二元卟啉器件的最高记录,也为小分子卟啉锌给体材料的分子设计提供了新的思路. 上述小分子给体材料的结构示于图6中.

Fig.6 Molecule structures of ZS1—ZS31
1.3 卟啉锌类小分子受体材料
与基于卟啉锌结构的给体分子相比,其作为电子受体的材料设计研究虽进展有限,但也有个别尝试取得了优异的效果. 基于先前的分析,使用强吸电子单元对卟啉单元进行4个中位的取代以降低卟啉结构的电子密度与能级是构建小分子受体的最优策略.
苝二酰亚胺(PDI)作为一个具有大共轭平面、高电子亲和力、高迁移率以及低能级的水平的结构单元,在小分子受体的构建中被广泛使用. 2017年, Li等[29]使用PDI作为卟啉锌的取代基合成了具有3D结构的星形小分子ZA1,通过乙炔基与4个PDI单元相连的卟啉锌单元表现出更低的能级,同时由于PDI单元的大位阻空间结构抑制了分子间的自聚集,使得PBDB-T∶ZA1构成的共混膜具有良好的相分离结构,该活性层薄膜在300~850 nm的较宽吸收范围内表现出较强的光谱吸收,基于该活性层组成的器件取得了7.4%的高PCE. 这项成果对卟啉锌衍生物小分子作为受体材料的研究起到了极大的推进作用. 同年, Hadmojo等[30]报道了一种由卟啉锌核心连接两个PDI单元组成的A-D-A结构小分子受体ZA2,基于PTB7-Th∶ZA2的器件取得了5.25%的PCE,尽管不是当时器件效率的最高纪录,但ZA2表现出1.27 eV的低带隙以及更强的电荷转移能力,这也佐证了构建四吸电子单元取代小分子的合理性. 2018年, Li等[31]又设计了一种基于苯并噻二唑与罗丹宁吸电子单元的四取代卟啉锌小分子受体ZA3,基于PDPP5T∶ZA3的器件仅取得了1.9%的PCE,但ZA3作为一种由新型强吸电子单元构建的小分子受体,为后续的研究提供了宝贵经验. 为了近一步研究PID作为吸电子基团在小分子卟啉锌受体中的应用, 2020年, Peng等[32]设计了一种全卟啉锌小分子太阳电池,其中用以DPP为吸电子端基的小分子ZS32作为给体,分别用基于PDI二取代、二聚PDI二取代、 PDI四取代以及二聚PDI四取代的卟啉锌小分子ZA4~ZA7作为受体制备了太阳电池. 在给体相同的情况下,以PID四取代的ZS6作为受体的器件取得了PCE为2.57%、 FF为44.3%的最优器件性能. 虽然这4种器件的效率并不高,但这是全卟啉锌小分子太阳电池的首次尝试,为后续研究开拓了道路. 上述小分子受体材料的结构示于图7中.
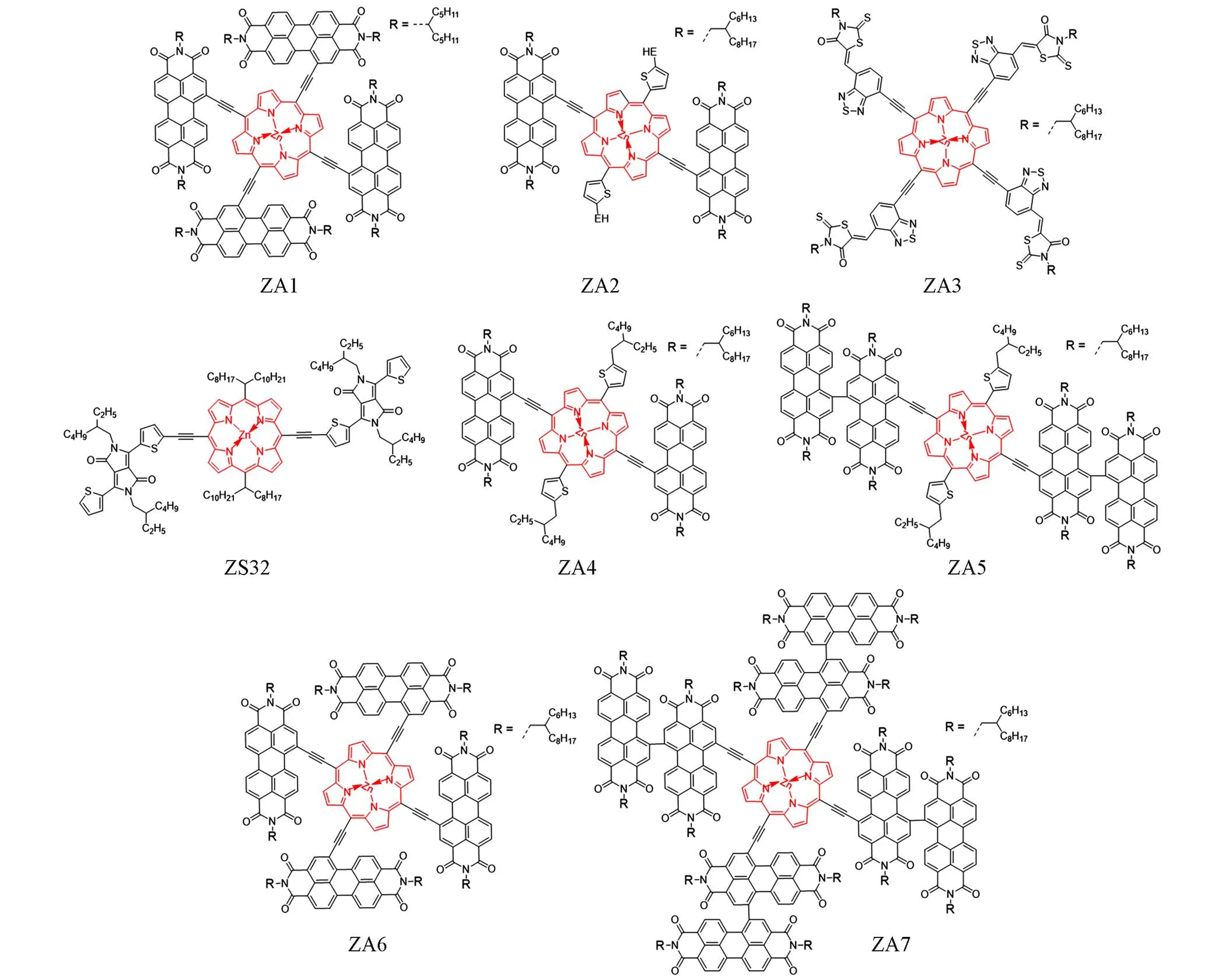
Fig.7 Molecule structures of ZA1—ZA7
2 铂基金属配合物材料
Pt(Ⅱ)炔金属类聚合物是研究最广泛的含Pt(Ⅱ)配合物的光伏材料之一,早在20世纪末,铂配合物就已经被应用于有机光伏领域. 半导体聚乙炔的诞生是导电聚合物领域的的一项开创性成果,而使用铂金属构建Pt(Ⅱ)炔类分子则为光伏材料带来独特的性质. 比如,铂作为一种具有较多电子的第Ⅷ族元素,与炔之间可以形成配键与反馈键,从而生成稳定的Pt(Ⅱ)炔配合物. 当Pt(Ⅱ)与炔基单元共轭形成一维聚合物链时, Pt(Ⅱ)的轨道与炔单元的轨道会互相重叠,从而导致电子离域并增强沿聚合共轭链方向的电子传播能力. 铂基金属配合物具有增强的自旋-轨道耦合,吸收光子形成的三线态激子具有更长的寿命,从而有利于增加激子的扩散长度. 同时相对于类似结构的分子,基于Pt(Ⅱ)的芳烃类化合物具有更好的溶解性,更有利于实现溶液加工电池器件的制备.
2.1 铂炔聚合物给体材料
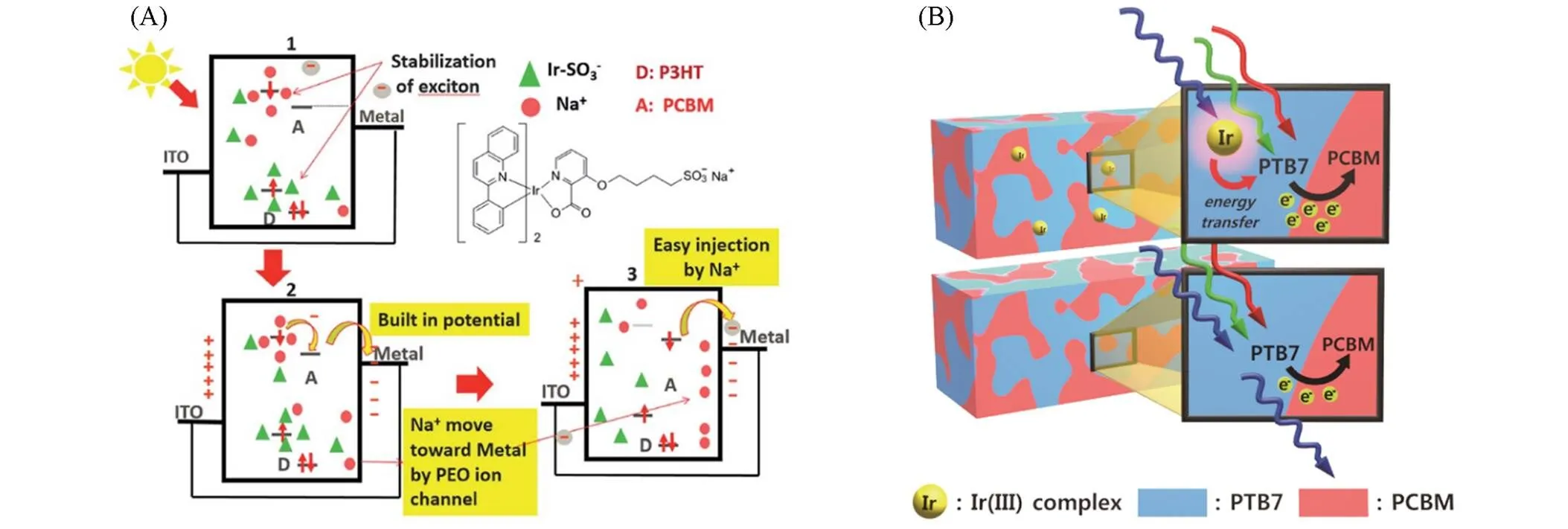
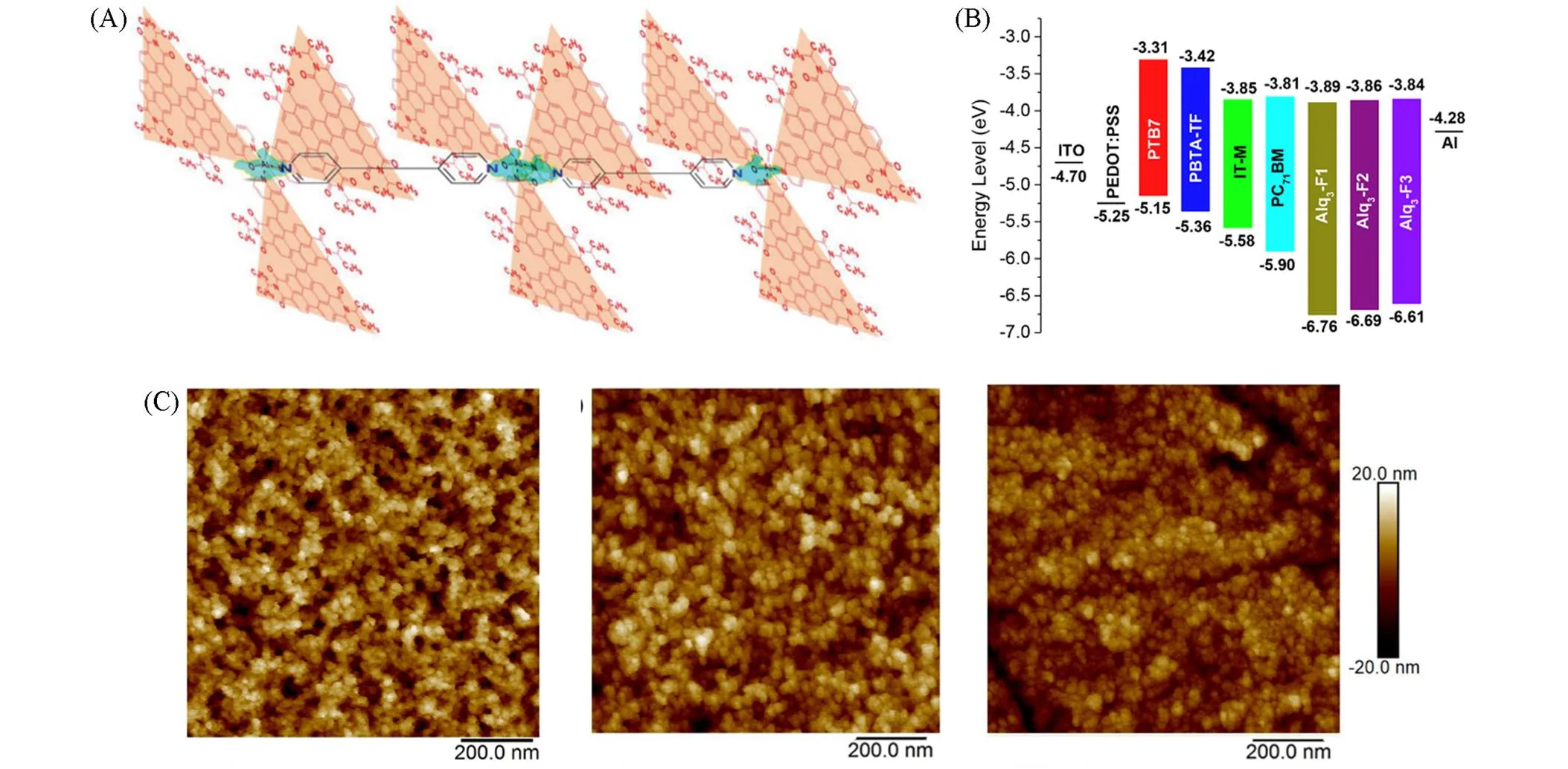
1994年, Köhler等[33]首次报道了基于苯间隔Pt(Ⅱ)炔的聚合物PP1,使用PP1与C60分别作为给体材料与受体材料时,实现了有效的电荷转移与分离. 对这种材料光致发光性能的研究表明,添加C60会降低分子磷光寿命,这说明三线态激子参与了电荷转移过程,基于ITO/PP1∶C60/Al的器件达到了1%~2%的光量子产率. 随后, Chawdhury等[34]制备了以寡聚噻吩为间隔的Pt(Ⅱ)炔的聚合物PP2~PP4,研究表明随噻吩数量的增多,聚合物的带隙(g)会随之减小. Reynold等[35]将PP2作为给体材料应用于OSCs中并观察到了光激发下三线态激子的产生与电荷转移,基于PP2∶PCBM的器件在活性层膜厚度为42 nm的条件下PCE达到了0.27%. 2007年, Wong等[36]将吸电子单元噻吩-苯并噻二唑作为间隔引入 Pt(Ⅱ)炔体系中,构建了基于D-A结构的窄带隙(g=1.85 eV)聚合物PP5,其带隙远低于以富电子单元苯作为间隔的PP1(g=2.55 eV). PP5具有更低的HOMO能级与更宽的吸收光谱,且在554 nm处表现出强烈吸收. 通过调整给、受体混合比或对使用的溶剂进行优化,基于PP5∶PCBM的器件最高PCE达到4.93%(oc=0.82 V,sc=13.1 mA/cm2). Wong等[37]利用3,4-乙烯二氧噻吩(EDOT)单元替换PP5中的噻吩单元合成了一种深蓝色聚合物PP6, PP6同样表现出了窄带隙和宽吸收范围,但由于较差的溶液处理性,基于PP6的太阳电池在未经优化时仅取得0.30%的PCE. Jen等[38]报道了一系列以苯并噻二唑与苯并二噻吩为间隔的聚合物PP7~PP9,该系列聚合物表现出了窄带隙(g=1.81~1.85 eV)以及高空穴迁移率(h=1.5×10‒3~1.0×10‒2cm·V‒1·s‒1),这归因于更具共轭与刚性的结构显著促进了沿聚合物主链 D-A单元的电子耦合,增强了ICT作用,同时引入烷基链以调控薄膜的非晶形态也是PP8与PP9获得高空穴迁移率的关键. 其中基于PP8∶PC71BM的器件获得了0.81 V的最高oc以及3.57%的PCE,而基于PP9∶PC71BM的器件获得了0.79 V的oc以及4.13%的最高PCE. 2011年, Wong等[39]报道了具有A--D--A结构间隔单元的聚合物PP10与PP11,三苯胺给电子单元与苯并噻二唑吸电子单元交替排列增强了分子内推-拉电子能力,使得分子在基态也能显著产生ICT效应. 其中,以噻吩作为桥的PP11的吸收范围在300~700 nm,并且拥有1.85 eV的窄光学带隙,基于PP11的器件获得了1.61%的PCE. Tzu等[40]与Wong等[41]基于PP5聚合物,引入了不同吸电子单元取代苯并噻二唑单元,合成了一系列光学带隙在1.47~1.97 eV范围的Pt(Ⅱ)炔类聚合物PP12~PP17. 基于这些聚合物的电池器件的oc在0.39~0.66 V之间. 通过调节不同吸电子能力的间隔中心与烷基链可以获得300~900 nm可见光至近红外区域的吸收覆盖. 这些器件的sc在0.25~2.99 mA/cm2之间, FF在0.17~0.38之间,低的sc和FF主要是由较差的激子分离和传输速度导致. 在这些聚合物给体制备的器件中,基于PP12的器件取得了0.68%的最高PCE,基于PP16的器件次之(PCE=0.56%). 随后, Wong等[42]报道了DPP和异靛单元 作为吸电子中心单元的聚合物PP18和PP19. 具有共轭双环结构的DPP单元具有很强的-相互作用,且具有能形成氢键的极性羰基基团,可以显著降低材料的g. 这两种黑蓝色的聚合物光学带隙在1.58~1.70 eV之间,并且在可见光波段表现出强烈吸收,具有用作给体材料的潜力. 同年,该课题组[43]设计了一些具有弱给体强受体结构单元的聚合物PP20与PP21,并应用于制备近红外太阳电池. 两种聚合物分别采用苯并双噻二唑和连接有喹诺啉单元的苯并噻二唑作为强受体,采用芴单元作为弱给体. 这种D-A-D型间隔的特殊设计起到了预期的效果,聚合物PP20与PP21表现出了1.54 eV与1.65 eV的窄带隙,并且具有约为5.5 eV的低HOMO能级. 在光谱吸收方面,归因于PP20的受体单元更强的吸电子能力, PP20较PP21的吸收发生了29 nm的红移. 基于PP20的器件取得了1.02%的PCE,高于基于PP21的器件(PCE=0.78%).
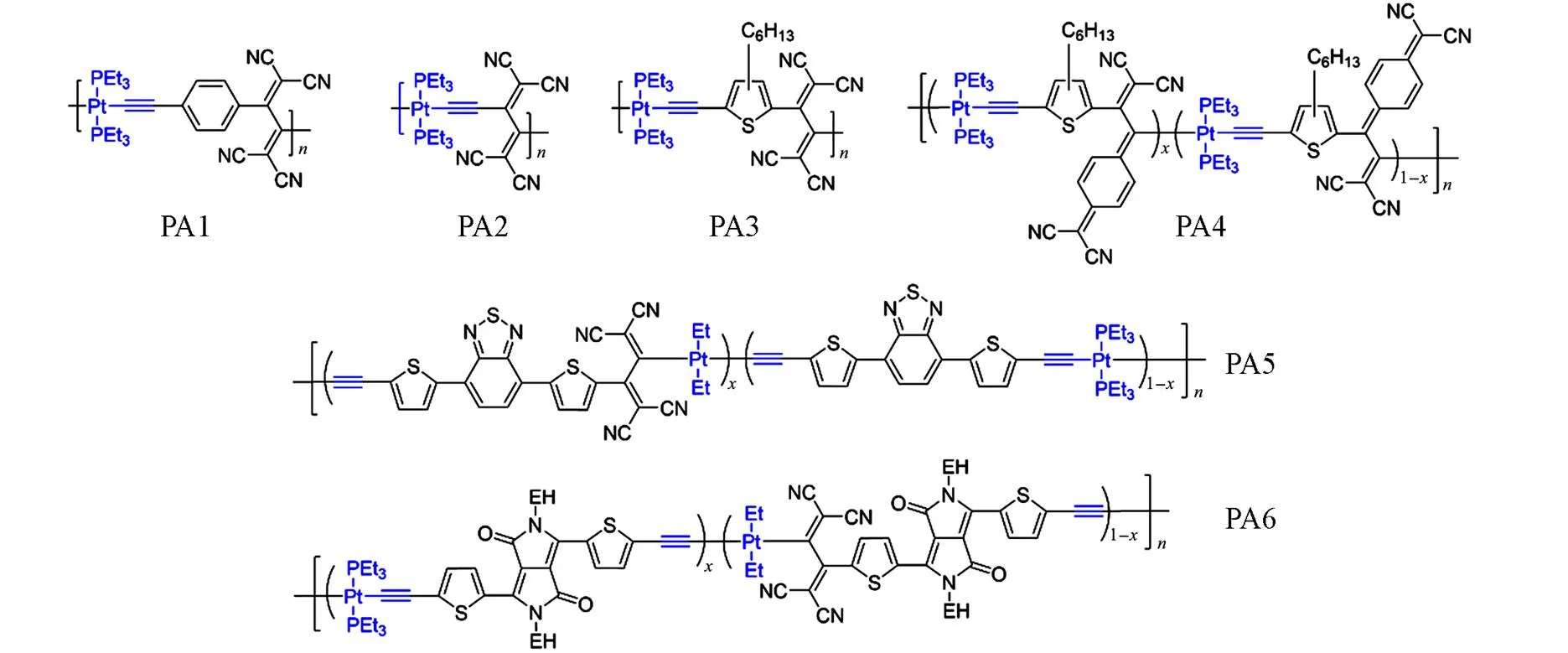
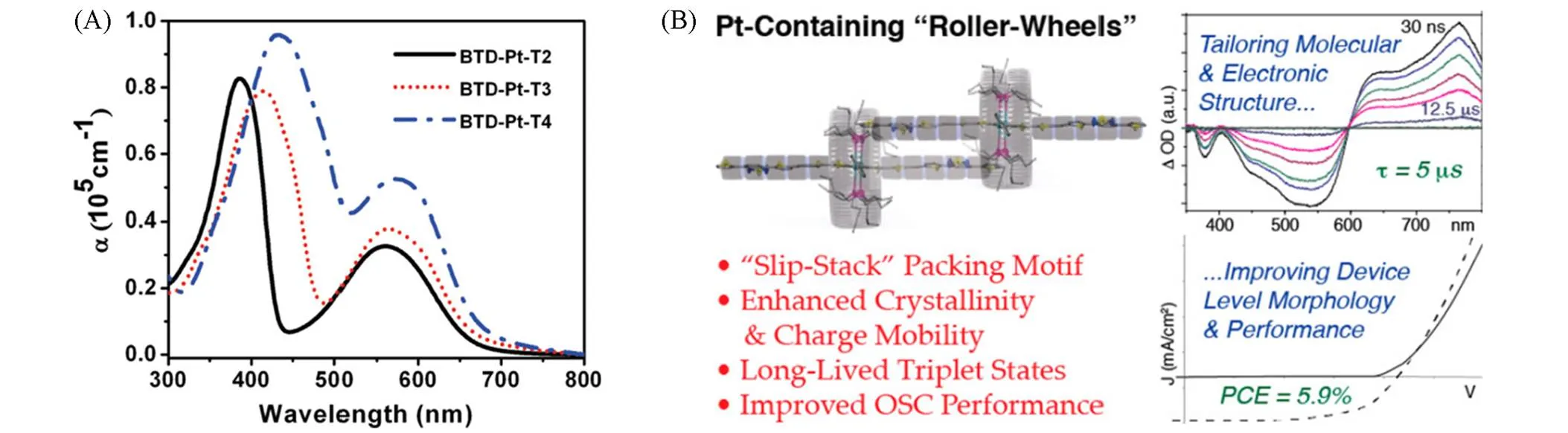
为了研究寡聚噻吩中噻吩数量对Pt(Ⅱ)炔聚合物的吸收、电荷传输等光电性能的影响, Wong等报道了使用不同噻吩个数的寡聚噻吩结构搭配多种吸电子单元的Pt(Ⅱ)炔聚合物. 在聚合物PP22~PP25[44]中使用了噻唑类强吸电子单元,通过测试发现,主链上噻吩单元个数的增加有效拓宽了聚合物的吸收光谱,空穴迁移率也随之增加. 噻吩数量的增多也会使得聚合物的带隙显著下降, PP25的g为2.19 eV,相比于没有噻吩的聚合物PP22带隙下降了0.27 eV. 基于噻吩数量较多的PP24与PP25的太阳电池分别获得了2.14%与2.5%的PCE,其余材料的PCE均低于1%. 在聚合物PP26~PP29[45]中使用芴作为吸电子受体,当由0增加到4时,聚合物的性质表现出了与PP22~PP25相同的变化趋势. 基于芴的铂炔聚合物的带隙较高(大于2.3 eV),但随着的增加,g从2.93 eV(PP26)降至2.33 eV(PP29). 尽管这类材料具有较宽的带隙,但由于具有较高的光吸收系数,该类材料与PCBM搭配时仍能产生较高的光电转化效率, PP26~PP29的PCE依次增大,基于PP29的器件取得了2.41%的最高PCE. 聚合物PP30~PP32[46]是使用强蓝光吸收的吩噻嗪发色团作为强给电子体,这些宽带隙的聚合物可用于短波长光照下的光敏化技术. 随主链上噻吩单元个数的增加, PP32表现出比PP31更强的吸收光谱,并且材料的电子和空穴迁移率都得到了提高. 这3种材料的PCE在1.03%~1.27%之间. 聚合物PP33与PP34[47]使用4-环戊并[2,1-b∶3,4-b′]二噻吩-4-酮作为间隔单元,这两种聚合物具有较低的带隙(1.44~1.53 eV),基于这两种聚合物的器件PCE均不足1%. 聚合物PP35与PP36[48]使用菲苯基咪唑作为侧链与主链噻吩相连作为间隔物,这两种材料表现出了2.28 eV与2.34 eV的宽带隙,但基于此材料的器件PCE均低于1%. 聚合物PP37与PP38[49]使用缺电子单元蒽醌作为 间隔物, PP38的带隙为1.88 eV,较PP37降低了0.42 eV,但基于PP38的器件PCE仅为0.35%. Wong等[50]在2011年还报道了一系列基于三苯胺的三维结构Pt(Ⅱ)炔类聚合物PP39~PP41,其空间结构如 图8(A)所示,作者认为具有三维各向同性结构的聚合物有望获得更好的光学性能. 与以噻吩作为臂的PP39相比,以芴单元作为臂的PP41具有更长的共轭结构和分子尺寸,因此PP41表现出了更好的吸收与电荷传输性能. 由于这3种聚合物不具备D-A结构,因此它们的光学带隙很宽(2.59~2.72 eV),随着三苯胺臂上Pt(Ⅱ)单元的增加,器件的开路电压从0.74 V提高到了0.82 V, FF也从0.38提高到了0.53,基于PP41的器件PCE达到了1.78%,高于基于PP39(PCE=0.83%)与PP40(PCE=1.6%)的器件. 上述聚合物给体材料的结构示于图9中.

Fig.8 Space filling model: top view(top) and side⁃view(bottom) of PP41[50](A), time⁃evolution of the fs⁃TAS spectra of PP48(left) and decay associated spectra stressing on the various transient species of PP48(right)[52], simulation of the self⁃assembled structure of the PP51 molecule[53](C)
(A) Copyright 2011, Wiley‐VCH Verlag GmbH & Co. KGaA, Weinheim;(B) Copyright 2020, the Royal Society of Chemistry;(C) Copyright 2021, American Chemical Society.

Fig.9 Molecule structures of PP1—PP41
Wong等[51]利用上述电子受体合成了一系列具有2个不同构筑单元的无规Pt(Ⅱ)炔共聚物PP42~PP47,由于存在两种D-A结构,这些聚合物显示出两个不同的ICT吸收带. PP42~PP44显示出了相似的-*与ICT吸收带, PP43与PP44的g几乎相同,但基于这3种材料制备的器件FF仅有0.2左右,这也导致基于PP44的最高PCE仅为0.71%. PP45~PP47为含有吡嗪结构受体单元的无规共聚物,它们的g几乎相同,在1.53~1.55 eV范围内,这说明噻吩并吡嗪结构对g起主要贡献作用,它们的PCE大小顺序为PP47(0.009%) Fig.10 Molecule structures of PP42—PP51 由于Pt(Ⅱ)炔聚合物固有的p型半导体特征,上述Pt(Ⅱ)炔聚合物均为OSCs中的给体材料,但在构建n型有机半导体Pt(Ⅱ)炔聚合物方面研究者也有许多的尝试. 通过将Pt(Ⅱ)炔化物与强吸电子基团,如四氰乙烯(TCNE)或7,7,8,8-四氰基醌二甲烷(TCNQ)等材料结合可以显著降低聚合物的能级,这体现了基于Pt(Ⅱ)炔化物制备OSC受体材料的潜力. 1981年, Sonogashira等[54]将TCNE引入聚Pt(Ⅱ)炔中制备了PA1和PA2,观察到此聚合物在可见-近红外区域显示出了明显的电荷转移(CT)吸收带. 随后, Michinobu等[55]使用己基噻吩单元开发了低带隙聚合物PA3和PA4,其带隙分别为1.83 eV与1.22 eV,两种聚合物都表现出了D-A特性,这导致ICT吸收带覆盖了近红外与可见光区域. 由于TCNQ更强的吸电子能力, PA4显示出了蓝移的CT带与更低的还原电位. Michinobu等[56]分别将BTD与DPP单元引入PA2中合成了聚合物PA5与PA6,它们的g分别为1.47 eV与1.28 eV, PA5与PA6与它们的前体PP5与PP18相比, LUMO能级有显著降低,说明使用环化加成使Pt(Ⅱ)炔氰基化是降低聚合带隙、拓宽吸收光谱的有效方法. 然而,基于PA5与PA6的器件显示出比基于相应的无氰基前体的器件更差的光伏参数,有研究者尝试制造以P3HT为给体、以PA5与PA6为受体的聚合物太阳电池,结果表明由PA5制成的器件仅产生0.00079%的PCE,而由PA6制成的器件甚至不产生任何光电流. 虽然Pt(Ⅱ)炔聚合物作为受体在OSC领域没有取得较好的成果,但这些尝试帮助研究者更系统地认识了光学和电化学带隙之间的关系. 上述聚合物受体材料的结构示于图11中. Fig.11 Molecule structures of PA1—PA6 Fréchet等[57]报道了以BDT为核心、寡聚噻吩为共轭臂的小分子PS1~PS3,这些分子表现出可见光区的强吸收与约1.9 eV的g[图12(A)]. 而随着共轭臂上噻吩数量的不断增多,小分子的吸收光谱发生红移,但对空穴迁移率的影响可以忽略不计. 基于这些分子的器件PCE为2.2%~3.0%,其中基于PS2的器件取得了3.0%的最佳光伏性能. Wong等[58]报道了一系列以Pt(Ⅱ)炔为核心、以两种不同端基构成A-D结构共轭臂的小分子给体PS4~PS7,通过引入不同的吸电子端基可以有效调节分子的吸收光谱与能级. 这些具有低HOMO能级的小分子与PCBM的LUMO能级可以很好地匹配,有利于获得高开路电压. 基于PS4, PS5, PS7制备的器件都达到了0.9 V以上的开路电压,且基于PS5∶PCBM(质量比 3∶7)的器件达到了1.59%的峰值PCE,基于PS7∶PCBM(体积比1∶4)的器件也获得1.56%的较高PCE,而由于PS6的成膜性不佳,基于PS6器件的光伏性能很差. Wong等[59]将BDT引入Pt(Ⅱ)炔小分子共轭臂,并以三苯胺作为端基构建了一系列具有A-D-A共轭臂结构的小分子PS8~PS10,并合成了无封端的小分子PS11作为对照. BDT端元与噻吩的良好共面性有助于改善材料的吸收性能,这些分子的吸收宽度均在600 nm以上. 由于三苯胺基团的给电子作用增强了分子的ICT,基于PS8~PS10的器件的光伏性能远高于基于PS11的器件,其中基于PS8与PS9的器件分别取得了7.10和7.15 mA/cm2的sc以及2.37%和2.34%的PCE. 2016年和2017年, He等[60,61]制备了具有滚轮形结构的新型Pt(Ⅱ)炔小分子PS12~PS14. 作者认为传统的Pt(Ⅱ)炔小分子由于较高的刚性结构往往不能表现出较好的结晶性,铂配合物位于小分子两端,形成哑铃状结构,不利于分子之间形成有效的-堆积,而新型的具有“滚轮”几何形状的Pt-双乙炔小分子由于大位阻的铂炔结构将小分子锁定,以一种滑移堆叠的方式获得更高的结晶性能[其结构如图12(B)所示],显示出更强的分子间相互作用以及良好的全色吸收. 其中具有长共轭臂的PS12与PS13的g分别为1.97 eV与1.94 eV,低于PS14的2.53 eV. 器件的PCE对活性层的共混形态非常敏感,其中基于PS13的器件达到了5.6%的PCE,这是当时基于Pt(Ⅱ)炔金属小分子给体材料的最高效率; PS14较短的共轭臂难以实现有效的分子间堆叠,这导致了材料较差的结晶性与低光伏性能. 上述小分子给体材料的结构示于图13中. Fig.12 Absorption spectra of PS1—PS3 thin films after annealing at 70 ℃ for 30 min[57](A) and structure and photovoltaic performance of PS12[61](B) (A) Copyright 2010, American Chemical Society;(B) Copyright 2017, American Chemical Society. Fig.13 Molecule structures of PA1—PA6 与Pt(Ⅱ)炔聚合物相比,基于铂金属环化的聚合物会表现出更高的柔韧性,可以通过对螯合配体进行修饰来调节材料的光伏性能. 在常见的环化铂有机配体和聚合物骨架的组合中, P t通常与O和N原子配位,其中心轨道依然在电子的光诱导跃迁中起重要作用. 重金属的强自旋耦合(SOC)效应使产生三线态激子以提高太阳电池性能成为可能,因此这类材料的开发具有重要意义. 2009年, Fréchet等[62]报道了两种环化铂聚合物CP1与CP2,使用Pt与2-(2′-噻吩)噻唑及二酮衍生物配体进行环化. 作者使用噻吩和芴作为不同的共聚单体,两种聚合物在可见光波段有很宽的吸收范围,分别显示出2.1和1.65 eV的光学带隙. 由两种聚合物材料CP2和CP1与PCBM制备成活性层的太阳电池分别显示出1.29%和0.4%的PCE,这表明环化铂聚合物在光伏材料的应用方向具有很大的潜力. 2013年, Cheng等[63]合成了两种环化铂聚合物CP3和CP4并与两种不含Pt的相同结构聚合物进行对比. 研究发现与没有铂的聚合物相比,环铂聚合物CP3和CP4表现出明显的红移吸收光谱和更低的带隙. IID作为典型的吸电子单元在构建D-A共聚物方面有很大优势, Schanze等[64]将环化铂配合物单体与具有刚性共轭平面的异靛(IID)衍生物共聚合成了聚合物CP5,聚合物的吸收光谱发生了红移. 然而,由于该材料的三线态与单线态能级十分接近,且三线态能级比电荷分离态能级低至少0.5 eV,导致三线态激子无法分离并快速发生非辐射复合,因此基于CP5的太阳电池仅取得0.22%的PCE. 该课题组[65]随后使用DPP替代IID单元, Pt分别与2-硫吡啶或2-苯吡啶成键制备了聚合物CP6和CP7. DPP单元显著拓宽了聚合物的吸收,其中CP6具有1.4 eV的带隙,起始吸收波长接近900 nm. 作者通过纳秒和皮秒瞬态吸收光谱检测到所含发色团产生的三线态激子[图14(A)]. 与CP7相比, CP6的载流子迁移率更高,以聚合物CP6为给体与PCBM制备的太阳电池FF约为66%, PCE为1.66%. 2021年,作者[66]将BDT单元引入环化铂聚合物的共轭骨架中制备了CP8. 但同样由于其三线态能级低于电荷转移态能级,导致三线态激子无法有效解离,器件的PCE只有1%. 在CP5与CP6研究的基础上, Schanze等[64,67]除了环化铂聚合物还尝试了基于相同结构的寡聚物CP9~CP11. 研究表明,基于环化铂的寡聚物比基于Pt(Ⅱ)炔的寡聚物具有更高的强自旋耦合效应,但CP9~CP11几乎没有任何光伏效应. Fig.14 Picosecond(left) and nanosecond(right) transient absorption spectra of CP7(steady⁃state absorption spectra for both polymers)[65](A) and 2D⁃GIWAXS patterns of the polymers CP13⁃n[69](B) (A) Copyright 2017, American Chemical Society;(B) Copyright 2019, Wiley‐VCH Verlag GmbH & Co. KGaA, Weinheim. 在高性能聚合物给体中引入适量环化铂配合物单元是进一步提升其性能的有效方法之一. 2017年, Tao等[68]将Pt(摩尔分数0, 1%, 1.5%, 5%)作为第三组分,通过环金属化与主链上的2-苯吡啶进行络合制备了聚合物CP12-Pt0, CP12-Pt1, CP12-Pt1.5和CP12-Pt5. 在较低分子量条件下,基于该系列聚合物的太阳电池的PCE由CP12-Pt0的7.92%显著提高到CP12-Pt1.5的8.45%. CP12-Pt1.5的高PCE得益于其更高的空穴迁移率、最少的孪生复合以及更高效的激子分离等. 但进一步将Pt摩尔分数提高到5%会导致聚合物分子量降低,聚合物的堆积与聚集大大减少,从而阻碍了光伏活性层中的电荷运输,也使得最终PCE下降. 该研究结果表明,将环化铂作为第三组分引入到现有的高效共轭光伏聚合物中是提高有机太阳电池PCE的可行方法. 2019年, Peng等[69]也使用了同样的策略,通过向多氮杂环聚合物给体中引入大位阻铂配合物Pt(Ph)2(DMSO)2来调整聚合物结晶度与分子堆积,从而优化活性层的形貌. 作者报道了s-四嗪受体单元和BDT给体单元共筑的D-A结构聚合物给体CP13-Pt0, CP13-Pt5, CP13-Pt10和CP13-Pt15,其中Pt(摩尔分数0, 5%, 10%, 15%)为第三组分. 当引入Pt配合物时, CP13-Pt0表现出1.93 eV的的宽带隙与-5.5 e V的HOMO能级. 它们的原位掠入射广角X射线散射技术 (GIWAXS)测试结果如图14(B)所示,可以看到,随着Pt配合物含量的增加,衍射强度逐渐降低,由(101)方向的衍射峰从z=1.73降低到1.71 Å(1 Å=0.1 nm)可知-堆积距离减小,这表明配合物的引入增加了空间位阻,从而降低了分子的聚集性,使排列更加有序. 先前的研究表明[68], CP13-Pt0的强聚集性会导致较差的相分离结构,限制了器件性能的提高,其与Y6制备的器件显示出25.10 mA/cm2的sc, 64.9%的FF值以及13.03%的PCE,但引入一定量含Pt(Ph)2(DMSO)2大位阻配体的配合物会显著改善共混膜的相分离形貌,其中基于CP13-Pt10∶Y6器件的sc和FF分别提高到了26.45 mA/cm2和76.3%,获得了0.52 eV的低能量损失以及16.35%的高PCE. 上述分子结构示于图15中. Fig.15 Molecule structures of CP1—CP13, CPA1—CPA3 在基于铂配合物受体材料的研究中, Tao等在现有的高效聚合物受体PNDIT2中引入Pt配合物开展了一系列研究. 2017年,他们[70]将弱吸电子能力的刚性Pt配合物按照不同摩尔比替换PNDIT2中原有的强吸电子萘二酰亚胺合成了系列聚合物受体CPA1-Pt0~CPA1-Pt5,由于Pt配合物的引入,其熔点、结晶温度、 HOMO和LUMO能级都有所提高. 基于该系列聚合物制备的器件性能显示,在相同结构的倒置器件条件下, CPA1-Pt2拥有最高的PCE(4.51%),而对照组CPA1-Pt0的PCE仅为3.88%,这种提高可能归因于CPA1-Pt2更高的空穴和电子传输性能以及更有效的电荷分离过程. 2018年, Tao等[71]将以二苯甲酰甲烷配体为主链的铂配合物按照不同摩尔比替换PNDIT2中的萘二酰亚胺,合成了新的聚合物受体CPA2-Pt0~CPA2-Pt5. 当使用PTB7-Th作为聚合物给体时,所制备器件的效率随着Pt配合物含量的增加而变大,基于CPA2-Pt5的器件获得了4.99%的PCE;当使用另一种聚合物PBDB-T作为给体时,器件性能表现出了相同的变化趋势,基于CPA2-Pt5的器件的PCE更是从3.79%提高到6.18%. 这主要是由于Pt含量增加的聚合物中更有效的激子分离、更少的电荷重组以及更高的空穴和电子迁移率. 随后,该课题组[72]还采用Pt的其它配合物以相同的方法合成了聚合物CPA3-Pt0~CPA3-Pt5,使用PTB7-Th作为给体时同样表现出器件的效率随着Pt配合物含量的增加而变大的趋势, PCE从2.55%逐渐提高到2.89%,最终达到3.26%. 以上材料设计与器件性能研究充分展示了环化Pt类聚合物受体在OSCs领域的巨大潜力. 上述分子结构示于图15中. 重金属Ir具有强自旋轨道耦合,且基于Ir的金属配合物具有高三线态激子产率与特定的八面体结构,因此通过可控的方式引入特定的配体可以实现对配合物光电性质的微调,基于环化Ir配合物的小分子或聚合物在OSCs领域具有独特的应用. Holdcroft等[73]于2008年报道了含有Ir的配合物给体材料IrD1,在与PCBM共混制备器件后发现其EQE由未添加Ir受体的1.1%提高至10.3%,而且0.07%的PCE也是未添加Ir配合物的35倍. 作者认为器件性能的显著提升是由于三线态激子的产生所致,具有更长寿命的三线态激子提高了扩散距离. 2012年, Yu等[74]使用乙酰丙酮和叔丁基苯基苯并噻唑啉作为Ir的配体合成了宽带隙荧光材料IrD2,该配合物显示出了5.5 eV的低HOMO能级,这使得IrD2与C60制备的器件具有较高的oc(0.89 V),但由于IrD2具有较低的空穴迁移率,导致器件的FF与PCE难以提高,基于IrD2∶C60的器件仅取得了2.10%的PCE. 2013年, Li等[75]使用2-(2,4-二氟苯基)吡啶(dFppy)和氮杂苝衍生物作为配体合成了具有近红外吸收的Ir配合物IrD3, IrD3显示出强烈的金属-配体电荷转移(MLCT),并且由于dFppy降低了分子的HOMO能级,基于IrD3∶C60的器件获得了0.99 V的oc与2.85%的PCE. 2014年, Zhen等[76]设计合成了两种Ir配合物IrD4和IrD5来探究由氧甲基苯或三苯胺取代的配体对器件光伏性能的影响,作者制备了由该系列配合物与PCBM共筑活性层的太阳电池,结果表明基于IrD5的器件EQE值远高于IrD4器件的,基于IrD5未进行优化的器件拥有2.0%的PCE,高于基于IrD4器件的(PCE=1.2%). 这一研究表明配体的选择会直接影响金属配合物太阳电池的光电性能. 2019年, Zhang等[77]使用不同结构的配体 Ftbpa与FOtbpa合成了小分子给体IrD6与IrD7,在FOtbpa中氧原子的桥连使得配合物的LUMO与 HOMO能级都有所提高,这使得IrD7与IrD6相比具有更低的电荷转移态能量;使用FOtbpa作为配体可以有效减少和抑制辐射和非辐射复合,基于IrD7与PCBM制备的器件获得了更高的oc与PCE,分别为0.88 V与3.56%. 2019年, Tao等[78]针对配体对共混膜形貌的优化设计合成了具有高度共轭配体TBz的小分子IrD8,经过优化后TBz配体表现出了高度平面化的共轭,由于具有较强的MLCT效应,该配合物在溶液中在350~500 nm范围内表现出较强的光吸收. 作者使用GIWAXS对IrD8与PCBM形成的共混膜进行表征[图16(A)],发现TBz与Ir配合后形成的八面体三维不对称结构可以显著抑制PCBM的自聚,起到优化共混膜形貌以改善载流子的分离与传输的作用;因此,基于IrD8的器件达到了3.81%的高PCE. 为了进一步提高器件效率,作者通过旋涂p型半导体PDCBT制备了PDCBT/IrD8∶PC71BM双层结构的太阳电池[79],使得器件PCE由3.81%提高到了6.17%,这是基于Ir配合物小分子的OSCs一项新纪录. 上述小分子给体结构示于图17中. Fig.16 AFM height images and phase images of IrD8∶PC71BM and PDCBT/IrD8∶PC71BM[78](A) and AFM topography images of the photoactive layer films for PM6∶Y6⁃C2(top) and IrP3⁃1.5∶Y6⁃C2(bottom), the 2D GIWAXS profiles for the pristine Y6⁃C2 film(top), blend PM6∶Y6⁃C2 film(middle), and blend IrP3⁃1.5∶Y6⁃C2 film(bottom)[82](B) (A) Copyright 2019, the Royal Society of Chemistry;(B) Copyright2020, Wiley‐VCH Verlag GmbH & Co. KGaA, Weinheim. Fig.17 Molecule structures of IrD1—IrD8 虽然小分子铱配合物具有很高的三线态激子产率,但由于其较低的共轭性降低了对光子捕获的能力,导致小分子Ir配合物作为给体材料的应用受到了许多限制. 为了获得更好的光伏材料,将铱配合物以一定比例引入现有的高性能聚合物给体中是进一步提升其性能的方式. 2015年, Hunag等[80]将三配位的(dfppy)2Ir(dbm)引入到当时备受关注的聚合物给体材料PTB7中,合成了一系列不同Ir配合物摩尔分数的聚合物IrP1-(=0, 0.5, 1, 1.5, 2.5, 5). 他们发现,低含量的Ir配合物对原给体的吸收、能级和共混膜形貌仅有轻微影响. 而随着铱配合物引入比例的提高,以该聚合物为给体、 PCBM为受体的器件光伏性能有所提高,器件的PCE从未添加Ir配合物的6.64%提升至8.71%(摩尔分数1%的Ir配合物),这可归因于Ir配合物的引入使得激子由电荷转移态向三线态传递的复合损失减少. 2018年,该课题组[81]还将相同的Ir配合物引入到聚合物给体PTB7-TH中合成了一系列聚合物IrP2-(=0, 0.5, 1, 1.5),基于不含Ir的聚合物给体与PCBM制备的器件PCE为7.9%;而当Ir配合物引入比例达到1%时,聚合物给体具有更高的空穴迁移率、更少的双分子复合与更有效的电荷分离,使得最终器件的PCE显著提高至9.2%. 2020年, Min等[82]将(dfppy)2Ir(dbm)引入到高性能聚合物PM6中,设计合成了一系列聚合物材料IrP3-(=0, 0.5, 1, 2.5, 5). 差示扫描量热分析结果表明,引入大位阻配体的Ir络合物使得PM6在103 ℃出现了新的放热峰,这表明络合物的引入降低了聚合物分子骨架的平面性与刚性,从而抑制了分子间聚集与-作用. 该方法可以合理地调制PM6的分子堆积,以获得合适的纳米形貌、有效的电荷传输并抑制激子复合以减少能量损失,其共混膜的AFM与GIWAXS测试结果如 图16(B)所示. 优化后基于IrP3-1.5的器件获得了17.32%的PCE,高于基于PM6∶Y6-C2的器件(16.07%). 2021年, Wong等[83]通过替换Ir配合物上的配体并引入PM6中合成了新的系列聚合物 IrP4-(=0, 1, 3, 5),基于该系列聚合物与Y6制备的器件在共混膜形貌、载流子迁移率及光学吸收方面都有很大的改进,其中PCE从15.65%提升至16.71%. 作者还通过将该系列聚合物作为给体材料,将两种非富勒烯N3和ITIC-Th作为受体材料制备了三元有机太阳电池[84], ITIC-Th作为第三组分可以增强光子捕获能力,并在三元活性层内形态调节优化分子排列和相分离结构,使器件的PCE从16.27%提高到17.22%. 上述聚合物给体的结构示于图18中. Fig.18 Molecule structures of IrP1—IrP4 Ir配合物独特的八面体结构与高三线态激子产率使其在给体材料的应用方面有所成就,但在受体材料的应用中却一直难以取得突破, 2015年, Kumar等[85]将Ir配合物小分子IrS1用作受体材料与给体材料P3HT制备成太阳电池,虽然二者可以很好地实现能级匹配,取得了1.12 V的高oc,但由于较低的电流密度与FF值使得PCE难以提高. Tao等[86]用含有氰基绕丹宁的铱配合物IrS2和IrS3作为受体材料,与PBDB-T给体共混制备了非富勒烯有机太阳电池,其PCE为1.62%. 作者认为器件效率较低的主要原因是铱配合物的八面体结构限制了分子的紧密堆积,影响了电子的分离与传输. 因此,通过结构设计改善Ir配合物受体材料的分子堆积结构是发展高性能Ir配合物受体、提高材料光电性能的重要发展方向. 上述小分子受体的结构示于图19中. Fig.19 Molecule structures of IrS1—IrS3 and IrA1—IrA7 Ir配合物的八面体结构与优异的三线态性能使得这类配合物作为添加剂应用于OSC器件中可以通过优化共混膜形貌、提高三线态激子寿命等途径来有效提高器件的光伏性能. 2007年, Horng等[87]将以mppy为配体的Ir配合物IrA1作为活性层添加剂应用在有机无机杂化太阳电池P3HT∶CdSe活性层中. 实验结果表明,相比于没有添加IrA1的器件,加入质量分数10%的IrA1使得器件活性层的三线态产率提高了一个数量级,器件的光伏响应显著增强. 2016年, Kim等[88]设计了一种含有烷基磺酸钠吊坠的Ir配合物添加剂IrA2,并将其与作为离子传输通道的聚氧化乙烯(PEO)应用到了以P3HT∶PCBM为活性层的器件中,当添加剂质量分数达到1%时,器件的sc提高了近20%, PCE也由未添加IrA2时的1.6%提升至3.4%,作者认为IrA2具有能量供体的功能,可以通过Na+的运动来减少活性层与金属电极之间的能量势垒,从而起到优化器件性能的作用,其工作机理如图20(A)所示. 2016年, Kwon等[89]报道了4种具有不同荧光范围的Ir配合物添加剂IrA3~IrA6,这些配合物均具有较强的MLCT,将这些聚合物用作PTB7∶PCBM活性层添加剂后研究发现,由于配合物与活性层材料良好的相容性与能级匹配,这些配合物在OSCs中实现了有效的能量转移[图20(B)],同时可以保持理想的活性层形貌,改善光吸收和光电转换过程. 其中IrA4添加剂使得器件的sc从13.3 mA/cm2提升至16.1 mA/cm2,相应的PCE从7.37%提升至8.72%. Ir配合物也可应用于有机太阳电池界面层材料. 2021年, Pu等[90]报道了一种Ir配合物小分子IrA7用作电子传输层材料,将这种材料用作PM6∶Y6器件的阴极界面时,器件的PCE由14.57%提升至15.75%. 作者认为IrA7界面层与PM6形成了一个微型太阳电池,这增大了活性层界面面积并改善了激子的解离与电荷提取过程,同时可以促进界面偶极的形成. 众多研究表明, Ir配合物在与其它材料结合时具有很强的通用性,这为进一步利用Ir配合物提高OSC的光伏性能提供了新的思路. 上述分子结构示于图19中. Fig.20 Plausible mechanism of the PEO ion channel in P3HT and PCBM with IrA2 and PEO[88](A) and schematic of energy transfer from an Ir(III) complex to PTB7∶PCBM[89](B) (A) Copyright 2014, the Royal Society of Chemistry;(B) Copyright 2016, American Chemical Society. 在含金属配合物本体异质结有机太阳电池的设计理念中,由于三线态激子具有比单线态激子更有效的电荷转移,利用重金属原子的重原子效应诱导有机物三线态的产生是研究构效关系的基本思路. 近年来,基于铂配合物的OSCs已有大量的研究成果,基于Pt(Ⅱ)炔金属类或环化铂类的太阳电池已经具有超过13%的PCE,这反映了金属配合物在OSC中应用的巨大潜力. 研究人员认为在聚合物中使用Ru替代Pt在原则上会更有优势,因为以Ru的轨道作为HOMO能级与配体*轨道作为LUMO能级导致了吸收的红移,这使得分子具有更宽的吸收光谱. 2011年, Colombo等[91]首次报道了基于Ru炔单元的D-A结构小分子RP1,其中两个Ru炔单元之间由二噻吩苯并噻二唑桥接, RP1的HOMO与LUMO能级分别为-4.74 eV与-3.07 eV,且RP1具有良好的光吸收能力,但由于严重的相分离结构导致较差的共混膜形貌,基于RP1与PCBM的器件仅取得了0.1%的PCE. 2017年, Wong等[92]报道了一系列基于Ru炔单元的多生色团小分子RP2~RP5,该系列分子由Ru炔单元作为给电子核心,以三苯胺/噻吩和BT分别作为给体和受体单元构筑具有D-A结构的共轭臂,噻吩和BT的强平面共轭与推拉电子效应显著增强了分子内电荷转移能力,该系列聚合物的带隙在1.70~1.83 eV范围内,并在300~700 nm范围内表现出较强的光谱吸收[图21(A)]. 与基于Pt炔的小分子PS8~PS10相比,相同结构的Ru炔小分子的吸收光谱表现出明显的红移. 与PCBM制备成共混膜用于器件表征时,基于RP2~RP5的器件分别获得了0.66%, 0.14%, 0.14%和0.25%的PCE,其中0.66%是当时基于Ru配合物OSC的最高PCE. 上述小分子给体的结构示于图22中. Fig.21 Normalized absorption spectra of RP2⁃5 in CH2Cl2 at 298 K[92](A), UV⁃Vis absorption spectra of the mono⁃Ru⁃based(B), bis⁃Ru⁃based(C) and tris⁃Ru⁃based(D) systems in CHCl3 and solid films[94] and schematic diagram of PSC device fabricated using rGO/RP19 as an electron donor[96](E) (A) Copyright 2017, Elsevier B. V;(B—D) Copyright 2017, Elsevier B. V;(E) Copyright 2017, the authors. 除了乙炔基,多联吡啶也是基于Ru光学活性配合物的常见配体,如2-2′联吡啶和2,2∶6,2,2″-三吡啶(terpy)等. 多联吡啶系配体既是键给体又是键受体, N原子上的孤对电子能与金属空轨道形成键,配体的空*轨道又能和适当几何形状的金属离子的占有轨道(如轨道)形成反馈键,这样的体系具有很高的化学稳定性,并使吸收波长红移. 2012年, Lin等[93]分别使用咔唑、芴、 BT作为吸电子单元与以三联吡啶作为配体的Ru配合物聚合合成RP6与RP7,这两种聚合物在260~750 nm范围内有较强的吸收,光学带隙在1.57~1.77 eV之间,基于PCBM制备的器件PCE分别为0.57%与0.9%. 2014年, Lin等[94]又报道了一系列Ru基树枝状超分子聚合物,分别包括单钌超分子RP8~RP10、具有吸电子单元核心的双钌超分子RP11~RP13以及具有给电子单元核心的三钌超分子RP14~RP16,这些钌基超分子覆盖了250~750 nm的广泛吸收,光学带隙为1.51~1.86 eV. 由于噻吩树枝状臂具有较强的给电子能力,更高的枝化度会导致金属配合物的HOMO能级升高,因此分子的吸收能力与光学带隙可以通过改变噻吩共轭臂枝化度进行有效调节,随着枝化程度的提高,分子的吸收能力增强,带隙显著降低,基于单钌、双钌、三钌的吸收光谱如图21(B)~(D)所示. 研究发现,基于BTD缺电子核心的双钌分子由于形成了推拉电子结构,可以取得更高的光伏性能,其中基于最大枝化度的双钌分子RP13与PCBM制备的太阳电池取得了0.77%的最高PCE,而基于三钌分子的器件PCE范围仅为0.06%~0.19%. 该作者[95]还曾报道过以三联吡啶作为配体的Ru配合物,主链含有二噻吩并环戊二烯和BTD单元的聚合物给体RP17和RP18,通过在RP17主链单元上引入氟原子得到的RP18具有更低的LUMO轨道和更窄的光学带隙,从而获得了更宽的吸收范围. 作者制备了以RP17/RP18为给体、 PCBM为受体的太阳电池,发现基于引入氟原子的RP18构筑的器件PCE由1.99%提升到2.66%,强电负性卤素原子的合理修饰显著提高了器件的光伏性能. Fig.22 Molecule structures of RP1—RP19 2017年, Neppolian等[96]通过共价功能化方法将含有以苯并咪唑为配体的钌聚合物接枝到二维还原氧化石墨烯(rGO)表面合成了聚合物RP19,其结构模型如图21(E)所示. 使用rGO作为支撑材料可以显著提高聚合物的机械强度,并且rGO具有优异的电学性能,可以实现电子的注入与迁移,而聚合物则起到了光采集天线的作用,瞬态光电流测量结果证明了rGO对于载流子分离起到了贡献. 当使用PCBM作为受体时,相比于参比器件,基于RP19的器件的oc与sc都得到了显著提高,最终获得了1.45%的PCE. 这种rGO与金属聚合物共价功能化的策略为新型光伏材料的设计提供了一种新的思路. 作者还尝试将RP19作为空穴传输层应用在倒置太阳电池器件中,结果发现基于ITO/PFN/PTB7∶PCBM/RP19/Al结构的器件取得了6.8%的PCE,显著高于使用PEDOT∶PSS作为空穴传输层的器件(PCE=5.1%). 上述聚合物给体的结构示于图22中. 随着研究人员对金属配合物光电性质中构效关系研究的不断深入,本领域已开发了众多性能优异、结构特殊的金属配合物并应用其特色功能来构筑高性能OSC器件,其中包括三(8-羟基喹啉)铝(Alq3),含乙酰丙酮(acac)配体的Zr, Os, Hf配合物等. 金属配合物Alq3及其衍生物已被广泛应用于有机发光二极管(OLEDs)中,研究表明Alq3具有很高的LUMO能级,是优异的传输电子-阻挡空穴的材料,这归因于相邻分子间存在有效的LUMO能级重叠,使得其对电子的传输能力是空穴的10倍以上. 2019年, Peng等[97]以Alq3为中心核, PDI或二聚PDI为外周基团合成了两个小分子受体Alq3-PDI与Alq3-PDI2,拥有大共轭平面PDI取代基的Al配合物形成了八面体结构,这样的结构显著增加了配合物小分子之间的距离,并且也抑制了PDI的自聚集作用. 实验结果表明,通过PDI和PDI2修饰可以有效降低Alq3的高LUMO能级,使得其能级可与PTTEA形成良好匹配. 同时,由于分子共轭平面增大和结晶度的改善, Alq3-PDI2的吸收较Alq3-PDI发生了明显的红移. 由于二聚PDI使得分子的吸收与共混膜的形貌均得到了改善,器件的PCE从基于Alq3-PDI的4.73%提升至基于Alq3-PDI2的6.33%. 随后作者使用4,4′-联吡啶(Bipy)作为活性层添加剂对基于Alq3-PDI2的器件进行优化, Bipy的氮可以和Alq3中心金属铝之间形成非共价键. Bipy分子锁不仅改善了基于Alq3的小分子的堆积,使得器件的PCE显著提高为9.54%,同时, Bipy可通过在共混膜中形成结构锁有效提高了器件的稳定性[图23(A)]. Fig.23 Schematic illustration of the molecular lock effect between Bipy and the Alq3 core for Alq3⁃PID2[97](A), energy level diagram of Alq3⁃F1—Alq3⁃F3 used in this work for fabricating PSCs[99](B), AFM height images of the TiAA films with the following annealing temperatures: low temperature(75 ℃, left), medium temperature(125 ℃, middle), and high temperature(200 ℃, right)[103](C) (A) Copyright 2019, Wiley-VCH Verlag GmbH & Co. KGaA;(B) Copyright 2018, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim;(C) Copyright 2019, American Chemical Society. 由于Alq3相邻分子间存在有效的LUMO轨道重叠,其LUMO能级通常位于‒2~‒3 eV的较高水平,且通常具有较高的电子传输能力,因此Alq3衍生物除了可以用作活性层材料外,亦可用作OSC器件中的界面层材料. 2005年, Song等[98]率先使用Alq3作为CuPc∶C60活性层器件的电子传输层,由于Alq3具有很好的水、氧气阻隔效果,使得在不影响原来PCE的情况下OSC器件的使用寿命提高了150倍以上. 2018年, Peng等[99]报道了3个基于Alq3衍生物的小分子电子传输层材料Alq3-F1~Alq3-F3,该系列材料以Alq3为核心,通过将溴化季铵盐的芴为共轭臂来实现材料的水/醇溶解性和低表面功函数(f). 研究表明随着共轭臂的延长,材料的f得到有效降低[图23(B)],但同时电子传输性能也随之降低;其中基于Alq3-F2的电子传输性能与f实现了最佳平衡,使得基于PTB7∶PC71BM的器件PCE达到了10.15%,基于PBTA-TF∶IT-M的器件PCE达到了13.75%,高于使用PFN-Br作为电子传输层的器件(PCE=12.24%). 在主链含金属-炔结构的聚合物给体的研究中, Huang等[100]在2013年报道了一种含有氨基化功能侧链的Hg炔聚合物PFEN-Hg,并将其应用在倒置OSC器件的电子传输层中. 得益于PFEN-Hg分子之间存在的Hg…Hg非共价作用以及炔基的-堆积作用, PFEN-Hg的电子传输能力大幅提升,因此基于PTB7∶PC71BM的器件PCE实现了由3.18%到9.11%的大幅提升. 同时作者发现氨基化功能吊坠具有显著的阻挡空穴的作用,防止了空穴电流在反向偏置时通过阴极泄漏,作者通过合成不含氨基结构的分子PFEN-Hg-a证明了这一点,使用PFEN-Hg-a作为电子传输层的器件PCE仅为5.98%. 含acac配体的金属配合物性质稳定且具有优异的水/醇溶性与电子传输能力,往往用作阴极缓冲层材料. 2014年, Tan等[101]使用醇溶性的乙酰丙酮锆[Zr(acac)4]作为电子传输层并将其应用于一系列基于聚合物给体与富勒烯受体的器件中. 实验结果表明, Zr(acac)4作为电子传输层可以与活性层形成良好的能级匹配来提取电子增强光电流,并且Zr(acac)4与Al阴极电极串联可以起到降低电阻的作用. 在基于PBDTBDD∶PC71BM的器件中,与使用Ca/Al阴极的器件相比,使用Zr(acac)4/Al作为阴极的器件PCE从7.34%上升到8.75%. 2017年, Tan等[102]报道了双乙酰丙酮二异丙基钛(TiPD)作为电子传输层在OSC中的应用,发现TiPD薄膜具有良好的光学性能,紫外-可见光范围内的透光率可达到90%,特别是对于倒置OSC的器件性能具有优异的效果,因此作者采用TiPD作为电子传输层,用于构建基于 PBDTBDD和ITIC-M的倒置器件,实验结果表明TiPD层可以与垂直相分离的光活性层相结合,有效提高器件中的电荷传输能力并抑制了载流子的复合,基于该器件的PCE为11.69%,与没有TiPD的倒置器件(PCE=6.13%)相比实现了91%的增强,这表明TiPD对于生产高效稳定的倒置OSC器件具有很大的研究价值. 2019年, Yang等[103]研究发现,通过对以TiPD为电子传输层的器件进行不同温度下的退火可以实现对TiPD分子结构的改变,从而对表面功函进行调整. 作者认为随着退火温度的升高, acac配体发生了分解, TiPD分子经历了LT TiAA(75 ℃)、 MT TiAA(125 ℃)、 HT TiAA(200 ℃)的结构变化 [图23(C)],这使得原有acac配体与中心金属Ti的键轨道重叠发生了改变,并影响了材料的功函,进而可以实现针对不同活性层的能级匹配来提高器件性能. 其中,基于该电子传输层与PBDB-T-SF∶IT-4F活性层的器件获得了13.2%的PCE. 2018年, Li等[104]报道了两种基于Sn的简单配合物Sn(acac)2和BuSnOOH,在基于PM6∶BTP-4Cl的器件中使用Sn(acac)2作为电子传输层使得PCE从13.75%增加至15.57%,而使用BuSnOOH作为电子传输层更是将PCE提升至17.01%. 作者通过实验证明了这些Sn基配合物附着在Ag电极上,可以显著降低其功函(4.47~4.06 eV),使其与受体的LUMO能级更加匹配. 与Sn(acac)2相比, BuSnOOH材料的薄膜形貌更加光滑,从而表现出更高的电子流动性和更高效的电荷提取能力,有效抑制器件中的孪生复合. 因此,使用BuSnOOH作为电子传输层时会表现出更高的器件性能. 2021年, Tan等[105]使用Hf作为acac配体的中心金属,通过在acac结构上引入苯环来不断扩展其共轭结构,合成了一系列聚集尺寸可控的配合物Hf(Acac)4, Hf(ACB1)4及Hf(ACB)4. 研究发现,随着配体结构上共轭程度的增加,金属配合物表现出了更强的分子聚集效应,使得Hf(ACB1)4与Hf(ACB)4在薄膜状态下形成比Hf(Acac)4更大的纳米颗粒[图24(A)和(B)]. 作者将这些配合物用作电子传输层,其可在光活性层表面形成分 布均匀的纳米粒子且具有光散射效应,提高光活性层的光捕获能力,从而优化器件性能. 以PM6∶ BTP-eC9为活性层、 Hf(ACB)4为电子传输层的器件PCE高达17.13%,且具有良好的稳定性. 该工作不仅展示了一种简单有效的途径来改变金属配合物的聚集尺寸,以获得合适的光散射中心,并且为设计一种简单、稳定、有效的电子传输层材料提供了新的思路. 2022年, Tan等[106]在先前Hf (ACB1)4的研究基础上对配体加以改进,设计并合成了一种配体上含有酰胺结构且具有自组装行为的金属配合物Hf (ACBN)4,酰胺结构显著提升了分子内与分子间氢键作用,氢键诱导的自组装行为使其表现出良好的溶剂抗性,可用作反向有机太阳电池中的电子传输层,而且在由SnO2/Hf (ACBN)4组成的界面层结构中, Hf (ACBN)4能有效填补SnO2表面的氧空位,钝化表面缺陷,实现更有效的电荷传输[图24(C)]. 活性层为PM6∶BTP-eC9∶PC61BM的器件实现了18.1%的高光电转化效率,此外, Hf(ACBN)4的紫外光过滤能力也显著提高了器件的光稳定性. 2022年, Liang等[107]报道了一种简便且低成本的乙酰丙酮Zn配合物,并将其与氨基氧化物功能化的苝二酰亚胺(PDINO)共混作为电子传输层. 结果表明,与原始的PDINO电子传输层相比,由于 PDINO和Zn(acac)2之间的电子转移,该材料表现出较低的电子亲和水平和更高电导率,因此基于复合界面层的PM6∶BTP-eC9器件可以实现PCE从16.4%到17.1%的提升,并且在界面层70 nm的厚度下仍可保持15.4%的高PCE,这些研究证明了乙酰丙酮金属配合物在构建低成本电子传输层材料方面的通用性与高效性. Fig.24 Plane⁃view TEM images of the spin⁃coated films of Hf(Acac)4, Hf(ACB1)4, Hf(ACB)4(A), ESP maps and the modeled bimolecular aggregation configurations of the three chelates[105](B) and initial and optimized configurations of SnO2/Hf(ACBN)4 with the corresponding binding energy values[106](C) (A, B) Copyright 2021, Chinese Chemical Society;(C) Copyright 2022, Wiley‐VCH GmbH. 此外, He等[108]将锇戊烷配合物与碳基材料相结合,通过改变取代基共轭长度制备了新型醇溶性金属纳米石墨烯材料HBC-H, HBC-P及HBC-S,并将其用作OSC器件中的电子传输层. 金属纳米石墨烯材料中更长的共轭链加强了分子内电荷转移,遏制载流子复合并使得分子间电子传输更为高效. 因此,随着分子共轭骨架的延伸,以PM6∶BTP-eC9为活性层的器件PCE从16.55%提高到18.05%,该研究表明锇金属-芳香体系具有较强的电子传输能力以及优化活性层和电极之间界面接触的潜力. 2021年, Peng等[109]利用低成本的Co(OAc)2·4H2O作为前驱体制备了一种新的钴基空穴传输层. 经过退火和紫外臭氧处理后,空穴传输层的功函数和电导率大大提高,降低了原有的形貌缺陷以及传输电阻. 在基于PM6∶L8-BO的器件中获得了18.77%的PCE,相比之下,以PEDOT∶PSS为空穴传输层的器件PCE为18.02%. 该研究为开发低成本、稳定高效的空穴输运层提供了新的思路. 上述金属配合物的结构示于图25中. Fig.25 Molecule structures of other metals 在有机分子框架中引入重金属原子,运用金属原子独特的性质来调控材料的物理化学性质从而改善材料的光伏性能,这为设计新型OSCs提供了新的思路. 本文基于不同金属的配合物在有机太阳电池活性层、界面层、添加剂等材料中的应用实例总结并分析了金属配合物结构与光伏性能之间的关系. 金属配合物在OSCs中的应用及推广方面依然面临许多挑战. 在活性层方面,即使添加了OMCs,高性能OSCs中依然是单线态激子起主要作用,三线态激子的应用十分有限;在界面层方面,基于OMCs空穴传输层的研究远落后于电子传输层的开发与器件应用, OMCs的低溶解性往往会增加溶剂化加工制备器件的难度,而且基于OMCs的界面层与活性层之间较难形成理想的欧姆接触等,这些局限性需要我们对OMCs的构效关系进行更深入的研究. 不可忽视的是, OMCs已取得众多好的结果,充分利用OMCs的优势可以为解决这些难题带来新的思路,作为一种可以通过修饰配体或改变金属离子来灵活调整其光电性质的材料, OMCs不仅可以改善共混膜的相分离形貌,调控分子能级,优化界面层光电特性,其较强的自旋轨道耦合可以产生大量具有更长寿命的三线态激子,从而提高激子扩散距离,而且OMCs作为一种具有较高稳定性的材料,可以解决传统界面层材料PEDOT∶PSS, MoO3, V2O5等制备困难、吸湿性高、酸度高的缺点,随着研究的不断开展, OMCs表现出了巨大的应用潜力. 在OMCs与其它光伏材料的相互作用机制尚未完全揭示的情况下,如何充分高效地利用OMCs的优点来调控器件的光伏性能仍是一个需要不断探索的问题,特别是在新型光伏材料不断涌现的今天, OMCs能否继续保持其优势而被广泛应用依然需要相关研究者的不断努力. 相信随着越来越多特殊设计的金属配合物的开发和应用,对其构效关系将产生更加深刻准确的认知,从而使OMCs材料能进一步加快有机太阳电池的商业化步伐. [1] Zhu L., Zhang M., Xu J., Li C., Yan J., Zhou G., Zhong W., Hao T., Song J., Xue X., Zhou Z., Zeng R., Zhu H., Chen C. C., MacKenzie R. C. I., Zou Y., Nelson J., Zhang Y., Sun Y., Liu F.,,2022,(6), 656—663 [2] Huang X. B., Zhu C. L., Zhang S. M., Li W. W., Guo Y. L., Zhan X. W., Liu Y. Q., Bo Z. S.,,2008,(19), 6895—6902 [3] Xiang N., Liu Y., Zhou W., Huang H., Guo X., Tan Z., Zhao B., Shen P., Tan S.,,2010,(5), 1084—1092 [4] Liu Y., Guo X., Xiang N., Zhao B., Huang H., Li H., Shen P., Tan S.,,2010,(6), 1140—1146 [5] Lee J. Y., Song H. J., Lee S. M., Lee J. H., Moon D. K.,,2011,(8), 1686—1693 [6] Zhan H., Lamare S., Ng A., Kenny T., Guernon H., Chan W. K., Djurišić A. B., Harvey P. D., Wong W. Y.,,2011,(13), 5155—5167 [7] Chao Y. H., Jheng J. F., Wu J. S., Wu K. Y., Peng H. H., Tsai M. C., Wang C. L., Hsiao Y. N., Wang C. L., Lin C. Y., Hsu C. S.,,2014,(30), 5205—5210 [8] Wang L., Shi S., Ma D., Chen S., Gao C., Wang M., Shi K., Li Y., Li X., Wang H.,,2014,(1), 287—296 [9] Wang L., Qiao Z., Gao C., Liu J., Zhang Z. G., Li X., Li Y., Wang H.,,2016,(10), 3723—3732 [10] Luo X., Wu F., Xiao H., Guo H., Liu Y., Tan S.,,2017,(2017), 205—211 [11] Bucher L., Tanguy L., Desbois N., Karsenti P. L., Harvey P. D., Gros C. P., Sharma G. D.,,2018,(1), 1700168 [12] Sharma G. D., Daphnomili D., Biswas S., Coutsolelos A. G.,,2013,(7), 1811—1819 [13] Huang Y., Li L., Peng X., Peng J., Cao Y.,,2012,(41), 21841—21844 [14] Li L., Huang Y., Peng J., Cao Y., Peng X.,,2013,(6), 2144—2150 [15] Xiao L., Liu C., Gao K., Yan Y., Peng J., Cao Y., Peng X.,,2015,(112), 92312—92317 [16] Liang T., Xiao L., Liu C., Gao K., Qin H., Cao Y., Peng X.,,2016,, 127—134 [17] Gao K., Miao J., Xiao L., Deng W., Kan Y., Liang T., Wang C., Huang F., Peng J., Cao Y., Liu F., Russell T. P., Wu H., Peng X.,,2016,(23), 4727—4733 [18] Li M., Gao K., Wan X., Zhang Q., Kan B., Xia R., Liu F., Yang X., Feng H., Ni W., Wang Y., Peng J., Zhang H., Liang Z., Yip H. L., Peng X., Cao Y., Chen Y.,,2016,(2), 85—90 [19] Cuesta V., Vartanian M., Pilar D. L. C., Singhal R., Sharma G. D., Langa F.,,2017,(3), 1057—1065 [20] Liang T., Xiao L., Gao K., Xu W., Peng X., Cao Y.,,2017,(8), 7131—7138 [21] Montcada N. F., Arrechea S., Molina—Ontoria A., Aljarilla A. I., de la Cruz P., Echegoyen L., Palomares E., Langa F.,,2016,, 330—336 [22] Chen S., Xiao L., Zhu X., Peng X., Wong W. K., Wong W. Y.,,2015,(77), 14439—14442 [23] Chen S., Yan L., Xiao L., Gao K., Tang W., Wang C., Zhu C., Wang X., Liu F., Peng X., Wong W. K., Zhu X.,,2017,(48), 25460—25468 [24] Aljarilla S. A. A., Pilar D. L. C., Palomares E., Sharma D. G., Langa E.,,2016,(41), 17953 — 17962 [25] Xiao L., Chen S., Gao K., Peng X., Liu F., Cao Y., Wong W. Y., Wong W. K., Zhu X.,,2016,(44), 30176—30183 [26] Aljarilla S. A. A., Cruz P. d. l., Singh M. K., Sharma G. D., Langa F.,,2017,(19), 4742 — 4751 [27] Lai T., Chen X., Xiao L., Zhang L., Liang T., Peng X., Cao Y.,2017,(37), 5113—5116 [28] Lai T., Xiao L., Deng K., Liang T., Chen X., Peng X., Cao Y.,,2018,(1), 668—675 [29] Zhang A., Li C., Yang F., Zhang J., Wang Z., Wei Z., Li W.,,2017,(10), 2694—2698 [30] Hadmojo W. T., Yim D., Aqoma H., Ryu D. Y., Shin T. J., Kim H. W., Hwang E., Jang W. D., Jung I. H., Jang S. Y.,,2017,(7), 5095—5100 [31] Guo Y., Zhang A., Li C., Li W., Zhu D.,,2018,(3), 371—373 [32] Pan X., Huang S., Zhu B., Xia R., Peng X.,,2020,(2020), 108503 [33] Köhler A., Wittmann H. F., Friend R. H., Khan M. S., Lewis J.,,1996,, 147—150 [34] Chawdhury N., Köhler A., Friend R. H., Wong W. Y., Lewis J., Younus M., Raithby P. R., Corcoran T. C., Al⁃Mandhary M. R. A., Khan M. S.,,1999,(10), 4963—4970 [35] Guo F., Kim Y. G., Reynolds J. R., Schanze K. S.,,2006, 17, 1887—1889 [36] Wong W. Y., Wang X. Z., He Z., Djurisic A. B., Yip C. T., Cheung K. Y., Wang H., Mak C. S., Chan W. K.,,2007,(7), 521—527 [37] Wong W. Y., Wang X., Zhang H. L., Cheung K. Y., Fung M. K., Djurišić A. B., Chan W. K.,,2008,(24), 3603—3612 [38] Baek N. S., Hau S. K., Yip H. L., Acton O., Chen K. S., Jen A. K. Y.,,2008,, 5734—5736 [39] Wang Q., Wong W. Y.,,2011,(2), 432—440 [40] Wu. P. T., Bull. T., Kim. F. S., Luscombe. C. K., Jenekhe. S. A.,,2009,, 671—681 [41] Wang X. Z., Ho C. L., Yan L., Chen X., Chen X., Cheung K. Y., Wong W. Y.,,2010,(3), 478—487 [42] Liu Q., Ho C. L., Lo Y. H., Li H., Wong W. Y.,,2014,(1), 159—168 [43] Qin C., Fu Y., Chui C. H., Kan C. W., Xie Z., Wang L., Wong W. Y.,,2011,(18), 1472—1477 [44] Wong W. Y., Wang X. Z., He Z., Chan K. K., Djurišić A. B., Cheung K. Y., Yip C. T., Ng A. M. C., Xi Y. Y., Mak C. S. K., Chan W. K.,,2007,(46), 14372—14380 [45] Liu L., Ho C. L., Wong W. Y., Cheung K. Y., Fung M. K., Lam W. T., Djurišić A. B., Chan W. K.,,2008,(18), 2824—2833 [46] Wong W. Y., Chow W. C., Cheung K. Y., Fung M. K., Djurišić A. B., Chan W. K.,,2009,(17), 2717—2726 [47] Wang X. Z., Wang Q., Yan L., Wong W. Y., Cheung K. Y., Ng A., Djurisic A. B., Chan W. K.,,2010,(9/10), 861—867 [48] Zhan H., Wong W. Y., Ng A., Djurišić A. B., Chan W. K.,,2011,(25), 4112—4120 [49] Li L., Chow W. C., Wong W. Y., Chui C. H., Wong R. S. M.,,2011,(6), 1189—1197 [50] Wang Q., He Z., Wild A., Wu H., Cao Y., U S. S., Chui C. H., Wong W. Y.,,2011,(7), 1766—1777 [51] Wu P. T., Bull T., Kim F. S., Luscombe C. K., Jenekhe S. A.,,2009,(2009), 671—681 [52] Nos M., Marineau⁃Plante G., Gao D., Durandetti M., Hardouin J., Karsenti P. L., Gupta G., Sharma G. D., Harvey P. D., Lemouchi C., Le Pluart L.,,2020,(7), 2363—2380 [53] Marineau⁃Plante G., Nos M., Gao D., Durandetti M., Hardouin J., Karsenti P. L., Lemouchi C., Le Pluart L., Sharma G. D., Harvey P. D.,,2021,(2), 1087—1096 [54] Takahashi S., Morimoto H., Takai Y., Sonogashira K., Hagihara N.,,2007,(2/3), 101—105 [55] Yuan Y., Michinobu T.,,2012,(20), 2114—2119 [56] Yuan Y., Michinobu T., Oguma J., Kato T., Miyake K.,,2013,(13), 1465—1472 [57] Zhao X., Piliego C., Kim B., Poulsen D. A., Ma B., Unruh D. A., Fréchet J. M. J.,,2010,(7), 2325—2332 [58] Cui C., Zhang Y., Choy W. C. H., Li H., Wong W. Y.,,2015,(2), 347—356 [59] Dai F. R., Zhan H. M., Liu Q., Fu Y. Y., Li J. H., Wang Q. W., Xie Z., Wang L., Yan F., Wong W. Y.,,2012,(5), 1502—1511 [60] He W., Livshits M. Y., Dickie D. A., Yang J., Quinnett R., Rack J. J., Wu Q., Qin Y.,,2016,(9), 5798—5804 [61] He W., Livshits M. Y., Dickie D. A., Zhang Z., Mejiaortega L. E., Rack J. J., Wu Q., Qin Y.,,2017,(40), 14109—14119 [62] Clem T. A., Kavulak D. F. J., Westling E. J., Fréchet J. M. J.,,2009,(6), 1977—1987 [63] Liao C. Y., Chen C. P., Chang C. C., Hwang G. W., Chou H. H., Cheng C. H.,,2013,, 111—119 [64] Goswami S., Gish M. K., Wang J., Winkel R. W., Papanikolas J. M., Schanze K. S.,,2015,(48), 26828—26838 [65] Goswami S., Hernandez J. L., Gish M. K., Wang J., Kim B., Laudari A. P., Guha S., Papanikolas J. M., Reynolds J. R., Schanze K. S.,,2017,(19), 8449—8461 [66] Holt E. D., Wang J., Winkel R. W., Younus M., Schanze K. S.,,2021,(2021), 100060 [67] Goswami S., Winkel R. W., Alarousu E., Ghiviriga I., Mohammed O. F., Schanze K. S.,,2014,(50), 11735—11743 [68] Wan Z., Yang J., Liu Y., Wang S., Zhong Y., Li C., Zhang Z., Xing G., Huettner S., Tao Y., Li Y., Huang W.,,2017,(32), 4729—4737 [69] Xu X., Feng K., Bi Z., Ma W., Zhang G., Peng Q.,,2019,(29), 1901872 [70] Gao X., Wang M., Cao X., Yang J., Zhong Y., Zhang Z., Li C., Huettner S., Tao Y., Li Y., Huang W.,,2018,(1), 105—115 [71] Gao X., Shi D., Wang M., Xue Z., Hu Y., Tao Y., Huang W.,,2018,(37), 9903—9913 [72] Gao X., Liang Y., Wang H., Yang T., Huettner S., Wang J., Zhu F., Tao Y.,,2019,(2019), 93—100 [73] Schulz G. L., Holdcroft S.,,2008,, 5351—5355 [74] Yu J., Zang Y., Li H., Huang J.,,2012,(21), 6653—6657 [75] Fleetham T. B., Wang Z., Li J.,,2013,(13), 7338—7343 [76] Zhen H., Hou Q., Li K., Ma Z., Fabiano S., Gao F., Zhang F.,,2014,(31), 12390—12396 [77] Jin Y., Xue J., Qiao J., Zhang F.,,2019,(47), 15049—15056 [78] Wu Q., Cheng Y., Xue Z., Gao X., Wang M., Yuan W., Huettner S., Wan S., Cao X., Tao Y., Huang W.,,2019,(18), 2640—2643 [79] Yang T., Gao X., He Y., Wang H., Tao Y.,,2020,(17), 5761—5768 [80] Qian M., Zhang R.,Hao J., Zhang W., Zhang Q., Wang J., Tao Y., Chen S., Fang J., Huang W.,,2015,(23), 3546—3552 [81] Xue Z., Wang S., Yang J., Zhong Y., Qian M., Li C., Zhang Z., Xing G., Huettner S., Tao Y., Li Y., Huang W.,,2018,, 1 [82] Sun R., Wang T., Luo Z., Hu Z., Huang F., Yang C., Min J.,,2020,(7), 2000156 [83] Zhang M., Ma X., Zhang H., Zhu L., Xu L., Zhang F., Tsang C. S., Lee L. Y. S., Woo H. Y., He Z., Wong W. Y.,,2022,(3)132832 [84] Zhang S., Zhang M., Wang X., Xu C., Xu W., Gao J., Wang J., Wong W. Y., Son J. H., Jeong S. Y., Woo H. Y., Zhang F.,,2021,(22), 5825—5832 [85] Baranoff E., Kumar P.,, University of Birmingham, John Wiley & Sons, Ltd., UK,2017, 655—680 [86] Yang T., He Y., Cheng Y., Gao X., Wu Y., Yuan W., Tao Y.,,2021,(28), 9871—9880 [87] Yang C. M., Wu C. H., Liao H. H., Lai K. Y., Cheng H. P., Horng S. F., Meng H. F., Shy J. T.,,2007,(13), 133509 [88] Yun M. H., Lee E., Lee W., Choi H., Lee B. R., Song M. H., Hong J. I., Kwon T. H., Kim J. Y.,,2014,(47), 10195—10200 [89] Kim H. T., Seo J. H., Ahn J. H., Baek M. J., Um H. D., Lee S., Roh D. H., Yum J. H., Shin T. J., Seo K., Kwon T. H.,,2016,(5), 991—999 [90] Zhou P., Liu Y., Gu J., Lian H., Lan W., Liao Y., Pu H., Wei B.,,2021,(19), 2100850 [91] Colombo A., Dragonetti C., Roberto D., Ugo R., Falciola L., Luzzati S., Kotowski D.,,2011,(6), 1279—1282 [92] Liu Q., Ho C. L., Zhu N., Fu Y., Xie Z., Wang L., Harvey P. D., Wong W. Y.,,2017,, 277—286 [93] Padhy H., Ramesh M., Patra D., Satapathy R., Pola M. K., Chu H. C., Chu C. W., Wei K. H., Lin H. C.,,2012,(6/7), 528—533 [94] Satapathy R., Ramesh M., Padhy H., Chiang I. H., Chu C. W., Wei K. H., Lin H. C.,,2014,(18), 5423—5435 [95] Feng K., Shen X., Li Y., He Y., Huang D., Peng Q.,⁃,2013,(23), 5701—5710 [96] Vinoth R., Babu S. G., Bharti V., Gupta V., Navaneethan M., Bhat S. V., Muthamizhchelvan C., Ramamurthy P. C., Sharma C., Aswal D. K., Hayakawa Y., Neppolian B.,,2017,, 43133 [97] Zhang G., Xu X., Lee Y. W., Woo H. Y., Li Y., Peng Q.,,2019,(29), 1902079 [98] Song Q. L., Li F. Y., Yang H., Wu H. R., Wang X. Z., Zhou W., Zhao J. M., Ding X. M., Huang C. H., Hou X. Y.,,2005,(1—3), 42—46 [99] Li Z., Xu X., Zhang G., Deng M., Li Y., Peng Q.,,2018,(11), 1800182 [100] Liu S., Zhang K., Lu J., Zhang J., Yip H. L., Huang F., Cao Y.,,2013,(41), 15326—15329 [101] Tan Z., Li S., Wang F., Qian D., Lin J., Hou J., Li Y.,,2014,(4691), srep04691 [102] Shi Z., Liu H., Wang Y., Li J., Bai Y., Wang F., Bian X., Hayat T., Alsaedi A., Tan Z.,,2017,(50), 43871—43879 [103] Wang R., Xue J., Meng D., Cheng P., Yuan J., Chang S. Y., Tan S., Jia B., Wang Z., Zou Y., Zhan X., Yang Y.,,2019,(34), 20800—20807 [104] Wang B., Chen X., Zhang Z., Zhang Y., Xiao C., Nu Y., Zhao C., Li W.,,2022,, 1566—1199 [105] Liu H., Yu R., Bai Y., Zeng Y., Yi Y., Lin J., Hou J., Tan Z. A.,,2021,(10), 37—49 [106] Yu R., Shi R., Liu H., Wu G., Ma Z., Gao H., He Z., Tan Z. A.,,2022,(31), 2201306 [107] Wang W., Lin Z., Li X., Tang Y., Zhong W., Zhang C., Yang T., Liang Y.,,2022,(2), 287—293 [108] Liu L., Chen S., Qu Y., Gao X., Han L., Lin Z., Yang L., Wang W., Zheng N., Liang Y., Tan Y., Xia H., He F.,,2021,(30), e2101279 [109] Meng H., Liao C., Deng M., Xu X., Yu L., Peng Q.,,2021,(41), 22554—22561 Recent Advances in the Application of Metal Complexes for Organic Solar Cells WANGJiarui, YURunnan, TANZhan’ao* (,,100029,) Organic solar cells have attracted much attention due to their advantages of light weight, diverse colors, and potential for fabricating flexible large-area devices. The innovation of donor and acceptor materials and interface layers is key to improving the performance of organic solar cells. Metal complexes, which combine the self-assembly order of complex molecules with the structural diversity of organic molecules, have high triplet exciton density and long exciton lifetime, having been regarded as an important photoelectric functional material. With the in-depth study of the photoelectric properties, various metal complex photoelectric materials have been successfully applied in organic solar cells to achieve higher power conversion efficiency. In this paper, the applications of metal complexes based on platinum, zinc, iridium, ruthenium, zirconium, and other metals as active layer material, interface layers, and additives of organic photovoltaic devices are reviewed, and the structure-performance relationships are analyzed in depth. In the end, the perspectives on the challenges and opportunities for these materials are prospected to provide reference and inspiration for the design and application of high-performing metal complexes in organic photovoltaic devices. Organic solar cell; Metal complex; Active layer material; Interface layer material O614;O631 A 10.7503/cjcu20230150 2023-03-30 网络首发日期: 2023-06-08. 联系人简介:谭占鳌, 男, 博士, 教授, 主要从事聚合物/钙钛矿太阳电池材料与器件和量子点/钙钛矿电致发光材料与器件方面的研究. E-mail: tanzhanao@mail.buct.edu.cn 国家自然科学基金(批准号: 21835006, 51873007)资助. Supported by the National Natural Science Foundation of China(Nos.21835006, 51873007). (Ed.: N, K, M)
2.2 铂炔聚合物受体材料
2.3 铂炔小分子给体材料

2.4 环化铂类聚合物给体材料


2.5 环化铂类聚合物受体
3 铱基金属配合物材料
3.1 铱基小分子给体


3.2 铱基聚合物给体
3.3 铱基小分子受体

3.4 铱基添加剂/界面层
4 钌基金属配合物材料
4.1 钌基小分子给体材料

4.2 钌基聚合物给体材料

5 其它金属配合物材料


6 总结与展望
