进化与大数据导向生物信息学在天然产物研究中的发展及应用
2023-09-16张凡忠相长君张骊駻
张凡忠,相长君,3,张骊駻
(1 西湖大学理学院化学系,浙江省功能分子精准合成重点实验室,浙江 杭州 310030; 2 浙江西湖高等研究院,理学研究所,浙江 杭州 310024; 3 复旦大学化学系,上海 200243)
进化是促进生命出现、发展和多样化的过程[1]。生命的进化本质上是基因信息的进化,而天然产物生物合成相关的酶由基因信息编码,因此也离不开进化的力量[1‑3]。植物和微生物为适应自然环境创造出许多天然产物。过去一个多世纪里,天然产物作为先导分子在医药健康和农业生产中发挥着重要作用,包括已被用作药物造福人类的青霉素、红霉素以及万古霉素等。生物信息学的预测分析表明,迄今为止,只有3%的细菌来源天然产物被发现,即使是链霉菌这样被高度研究的类群仍然含有许多未知的天然产物[4]。
基因测序技术的发展带来基因组数据的快速增长(图1),大规模基因组学、代谢组学和功能研究的系统数据即“大数据”与生物信息学的结合为天然产物研究带来了“技术革命”。传统的天然产物研究高度依赖于化合物分离纯化,过去我们只能通过积累分离得到的单体化合物来了解天然产物。如今,我们已处在向天然产物全景图可视化过渡的阶段。基于大数据和生物信息学的现代天然产物研究使我们对天然产物的分子多样性、丰度以及分布有了宏观认识,不仅让我们了解到微生物天然产物库的巨大挖掘潜力,也可以指导具有临床或商业价值的新分子的发现,提高天然产物发现的效率[1‑2,5‑8]。由于已测序的真菌基因组数量相比细菌要少很多(截至2023 年1 月,NCBI数据库中细菌基因组数为1 420 776,而真菌为28 183,只占细菌基因组数量的2%),进行大数据分析主要以细菌为主,因此本文主要阐述细菌天然产物研究,但也包括了部分真菌研究。

图1 2002-2022年(近20年)NCBI数据库中细菌基因组数量的增长趋势Fig. 1 Growing trends for the number of bacterial genomes in the NCBI database from 2002 to 2022(in the last two decades)
对天然产物生物合成机理及相关酶的生化特性的认识促进了进化分析在酶功能预测方面的应用,进而指导酶的改造以及生物合成途径的改造[6,9]。目前基于进化进行的天然产物研究主要集中于以下几个方面:①利用进化导向方法发现新的天然产物(化合物结构预测);②通过进化分析预测酶的功能;③通过改造生物合成体系创造出我们需要的产物。因此,本文将聚焦于基于大数据的进化导向生物信息学方法在天然产物发现和酶工程研究中的运用进展,并对这些工具、方法在天然产物研究领域的发展进行展望。
1 基于进化和大数据的天然产物研究策略
基因挖掘是在没有化学结构的前提下,基于遗传信息来预测和分离活性天然产物[10‑13]。微生物基因组挖掘方法振兴了抗生素的研究,但这些方法依赖于先前识别的生物合成酶的序列相似性搜索,这种经验性质限制了所探索的化学空间。近年来,天然产物研究人员通过将进化原理融入基因组分析来寻找新的路径[14‑15]。利用进化和大数据进行的天然产物研究一方面是基于系统发育距离预测功能相似度:当靶标蛋白序列与已知化合物的编码序列距离较远且形成不同进化分支时,倾向于产生具有新核心结构的新产物;当靶标序列与已知化合物编码序列相邻时,可能会产生与已知化合物变化不大的新产物[图2(a)][10,15]。另一方面可以依据目标序列是否与已知序列聚集在一起,或属于一个新的分支,或是一个罕见的异常值,实现分布模式和多样性的可视化,进而展示一个全景图[图2(b)]。
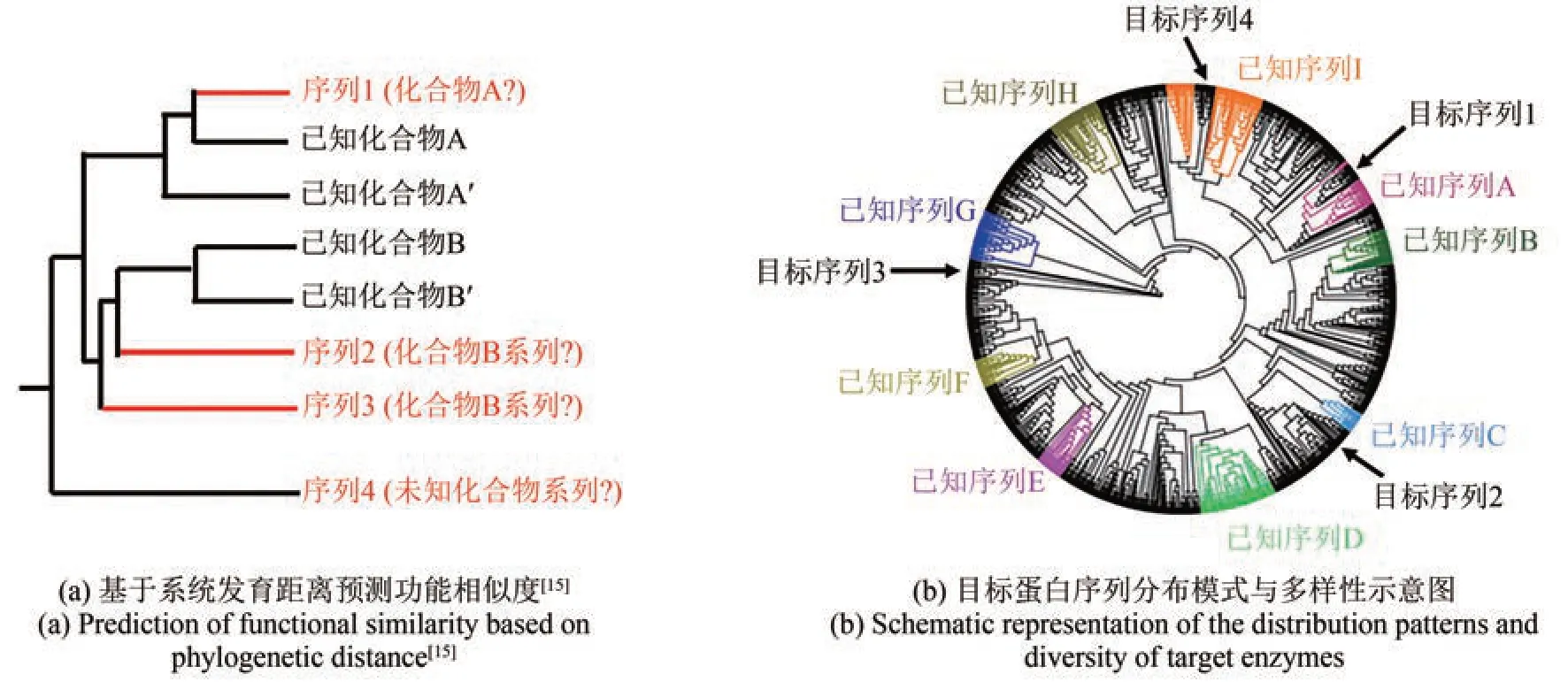
图2 进化分析的核心观念Fig. 2 A core concept of the phylogenetic analysis
已有不少基于进化的生物信息学工具可用于天然产物的挖掘,例如ARTS(Antibiotic Resistant Target Seeker)[16‑17]、 NaPDoS (Natural Product Domain Seeker)/NaPDoS2[18‑19]、 EvoMining[20]、Big‑SCAPE 和 CORASON[21‑22]等。 ARTS 和EvoMining 面向进化上相关的基因组,针对保守的生物合成基因簇(biosynthetic gene cluster, BGC)进行预测和聚类分析。其中ARTS以抗性基因为靶标,通过将管家基因、已知抗性基因与邻近BGC联系起来,自动筛选序列数据,挖掘作用方式新颖的抗生素,还可以用于比较相似的BGC 及其假定的抗性基因;而Evomining 基于酶的基因复制和底物特异性扩展的进化方式开发了一种基因挖掘方法,可以检测某些类型的管家基因的同源基因,并比较每个酶家族的平均数量和系统发育距离,因此可以用于直观地展示天然产物生物合成酶的起源与进化方向。NaPDoS是将PCR 产物、基因组或宏基因组数据快速提取和分组,分析目标KS(ketosynthase)或者C(condensation)结构域在进化树上的位置,从细菌遗传数据中推断次生代谢物的新颖性和潜力。Big‑SCAPE 是面向进化信息未知的多个基因组,以MIBiG 数据库[23]的基因簇作为参考分析antiSMASH[24]预测的基因簇,构建序列相似性网络,并把这些基因簇分为不同的基因簇家族,然后用CORASON 解释每个基因簇家族中不同基因簇的进化关系。这些生物信息学的工具同是基于进化原理,但是可以针对不同类型的基因以及用于实现不同的目的。
目前利用进化方法研究得最好的一类天然产物合成酶是模块型酶,如聚酮合酶(polyketide synthase, PKS) 和非核糖体肽合成酶(non‑ribosomal peptide synthetase, NRPS)。根据目标基因簇类型,我们对进化导向研究的进展进行了分类讨论,主要包括PKS、NRPS 以及其他非模块型酶。
2 进化和大数据导向的PKS研究
聚酮化合物是一大类具有广泛结构和功能的生物活性天然产物,临床上使用的多种药物都属于聚酮类,例如红霉素、阿维菌素、四环素等。聚酮化合物由聚酮合酶(PKS)负责生物合成。目前已知的细菌的PKS有3类(Ⅰ型、Ⅱ型和Ⅲ型)。其中,Ⅰ型PKS 由多个模块形成组装线,每个模块都包含核心结构域KS、AT(acyltransferase)、ACP(acyl carrier protein)协同催化聚酮链延伸的一个循环,部分模块含有KR(ketoreductase)、DH(dehydratase)、ER(enoylreductase)等结构域对聚酮进行不同程度的修饰[25][图3(a)]。Ⅱ型PKS 通过聚酮合酶(KS/KSα)和链长因子(CLF/KSβ)催化乙酸单元的迭代缩合反应,随后通过还原、环化和脱水反应形成多环芳香族骨架[图3(b)][26]。Ⅲ型PKS,也称查尔酮合成酶样PKS,属于同型二聚体酶,本质上是迭代作用的缩合酶[27‑28]。许多研究者尝试从进化的角度去理解自然界中PKS 基因与聚酮化合物结构之间的关系,主要是由于:首先,对天然PKS 多样性的探索有可能发现新的生物活性聚酮;其次,PKS 的多模块结构也为研究多个同源但功能不同的蛋白进化提供了一个独特的例子;最后,更好地理解自然界聚酮多样化形成机制可以为PKS改造开辟新的途径[29]。

图3 PKS生物合成Fig. 3 Biosynthetic pathway of PKS
2.1 PKS的基因进化机制
通过对PKS 不同结构域(KS、AT 和KR 等)的进化分析,目前认为导致PKS 装配线多样化的进化过程主要包括基因复制、水平基因转移、基因转换以及重组[图4(a)][29]。Ⅰ型PKS的模块化几乎都来源于单个祖先模块的多个副本[30]。重复模块为基因重组提供了理想的平台,可以导致产物化学结构的相应变化[30]。除了基因重组,基因进化过程中DNA 序列可能从一个同源区域非交互地转移到另一个同源区域,从而使这些同源序列同质化,即发生基因转换。基因转换广泛存在于Ⅰ型PKS 中[31]。对PKS 的这些“自然重编程”事件的分析可能有助于开发生物活性化合物的生物组合设计[30]。

图4 PKS进化机理Fig. 4 Evolutionary mechanism of PKS
cis‑AT PKS的进化被认为主要通过模块复制以及整个组装线的水平或垂直获取[32]。相反,trans‑AT 系统具有明显的重组和嵌合形成新的基因簇的趋势[33]。进化树上相近的trans‑AT PKS 的KS 往往催化相似结构的底物[33],而对于cis‑AT PKS,同一个基因簇的KS结构域往往具有高序列相似度[34]。Ikuro Abe 等通过对4 个氨基多醇(neomediomycin B、mediomycin、ECO‑02301、tetrafibricin)的基因簇分析发现KS 结构域表现出与上游模块的ACP更近的进化关系,这表明cis‑AT PKS 中KS 与上游模块的ACP 及修饰结构域作为整个单元进行重组,自然的重组发生在KS 与AT 或AT 与修饰结构域之间[35‑36]。Adrian Keatinge‑Clay 随后的分析表明trans‑AT PKS 的KS 结构域也表现出与上游模块的ACP 更近的进化关系[37]。这与KS 门控角色相符,因为KS 底物的结构由上游模块的组成所决定。这也形成了装配线PKS 模块的重新定义AT‑(DH‑KR‑ER)‑ACP‑KS,而非传统的KS‑AT‑(DH‑KR‑ER)‑ACP[图4(b)][36]。
不同于KS,KR结构域在系统发育树上基于产物羟基的立体构型分类[38],在trans‑AT PKS 中还依据其他修饰结构域的存在进行分类[39]。一种观点认为这是由于基因转换不需要影响PKS 模块的所有结构域,所以修饰结构域并不总是与KS 共进化[29]。这也就意味着,在模块内部同样存在着结构域自然重组的位点。我们注意到,导致遗传序列同质化的协同进化在PKS 这样的重复遗传区域经常发生[40],而这会掩盖进化轨迹。因此,要阐明PKS 的确切进化过程还需要进行仔细分析。
Ⅱ型PKS 基因簇的核心KS 和CLF 基因被认为来源于一个古老的KS复制[41],它们可能在放线菌诞生之前就从FabF(脂肪酸途径中的KS 同源蛋白)的共同祖先分化而来,然后在很少甚至没有基因交换的情况下共同进化[42]。与KS 树相比,CLF 有更清晰的分支结构,且CLF 的进化分支与聚酮结构单元数目(而非总碳数)的关系更紧密[5]。除了KS 和CLF 基因,Ⅱ型PKS 基因簇中的聚酮还原酶与环化酶被认为是从其他系统交换到基因簇中,随后进化出PKS特异性功能[42]。
2.2 PKS的功能预测
进化分析最重要的应用之一是区分旁系同源(由重复产生)和直系同源(由物种形成产生)的基因或蛋白。一般情况下,同源与相同的功能相关[43]。因此能够利用系统基因组方法从进化亲缘来预测序列功能,即对功能未知的基因根据它们相对于已知基因的系统发育位置进行功能预测。
对PKS 的功能预测目前主要针对KS 结构域,Ⅰ型trans‑AT PKS 的KS 结构域[30,32]、Ⅱ型PKS 的CLF 结构域分支[5,42]等均能形成与其底物的化学结构紧密相关的进化分支,因此可用于化合物结构预测以及同工酶的预测。KS 结构域序列不仅可以用来区分聚酮与脂肪酸、烯二炔和多不饱和脂肪酸,而且还可以用来区分不同类型的聚酮,如cis‑AT PKS 和trans‑AT PKS、PKS/NRPS 杂合型,NaPDoS 即是利用这一原理进行酶功能和化合物结构预测[18‑19]。此外,Ⅱ型PKS 中修饰酶的功能也可以通过进化分析预测,如KR 的区域选择性和环化酶的环化方式[42,44]。
除了根据已知功能的酶来推测同家族相似酶的底物和功能外,还可以依据基因的共同进化来研究未知酶。BGC 的发展经历了不同的进化过程,如基因组内的复制、重排、域/模块/基因交换和水平基因转移[30,40]。同一个簇中那些相互作用的酶需要共同进化的过程,以保持适当的相互作用[6]。因此还可以根据与之有相互作用的酶来预测未知酶的功能。Jorn Piel 课题组[45]通过对trans‑AT PKS 中KS 的进化分析,发现了一个包含TEB类(负责O‑乙酰化)结构域的KS 分支。根据产物结构及缺失的HGTGT 活性位点,推测这些KS 均为非延伸的KS0,尽管催化的聚酮结构不同,但TEB模块具有生物化学上的一致性。
细菌Ⅰ型PKS 中除了KS 结构域可以形成与底物化学结构密切相关的进化分支,AT 结构域在进化树上也形成两个主要分支,分别具有接收丙二酰‑CoA 和甲基丙二酰‑CoA 的特异性[46]。AT 结构域对于丙二酰‑CoA 和甲基丙二酰‑CoA 的特异性识别可以通过序列中两个特征区域预测,HAFH 和GHS(I/V)G 序列表明其接收丙二酰‑CoA,而YASH 和GHSQG 表明其接收甲基丙二酰‑CoA[47‑49],这一发现已长期被用于区分这些AT 的底物选择性。
2.3 PKS的基因挖掘
KS 结构域的进化分析用于化合物结构预测以及同工酶的预测是PKS 基因挖掘的理论基础。对芳香聚酮BGC 中的KSα和CLF(即KSβ)进行的进化分析表明,KSα和CLF 的进化树结构和分支模式都很相似,而且以对应聚酮化合物的链长聚类,因此KSα和CLF可作为理想的进化标签代表整个基因簇[42]。Sean Brady 课题组[50‑51]以CLF 序列作为进化标签,从土壤微生物组中扩增相关基因,与已知CLF 基因做序列比对并进行进化分析,发现了许多与已知序列在同一分支的不同亚支的序列,通过将对应的基因簇在菌株Streptomyces albusJ1074 中异源表达发现了结构新颖且活性显著的多酚和蒽环霉素类化合物(1~3,结构见图5)。最近,本课题组[5]在Ⅱ型PKS 中应用全局基因组挖掘,从一个与已知基因簇距离较远的进化分支上发现了oryzanaphthopyrans(4,结构见图5),此外,也基于大数据的进化分析描绘了Ⅱ型PKS 的分布、丰度以及多样性的全景图。戈惠明组[52]利用CLF 的进化模式结合抗性基因靶向挖掘四环素类化合物,发现了高度糖基化的四环素海南霉素(5,结构见图5)。这些研究或不需要培养微生物或在微生物培养前已能够预测化合物的结构新颖性和生物活性水平,体现了进化导向基因挖掘的优势。

图5 进化导向基因挖掘获得的聚酮化合物1~11分子结构化合物1~5为Ⅱ型PKS基因挖掘的芳香聚酮;化合物6~9为trans‑AT PKS基因挖掘产物;化合物10为真菌Ⅰ型PKS基因挖掘产物;化合物11为烯二炔类聚酮化合物Fig. 5 The structure of polyketides molecules 1 to 10 obtained by phylogeny‑guided genome miningCompounds 1 to 5 were aromatic polyketides discovered by genome mining of type Ⅱ PKS. Compounds 6 to 9 were discovered by genome mining of trans‑AT PKS. Compound 10 was discovered by genome mining of fungal type Ⅰ PKS. Compounds 11 were enediynes
在Ⅰ型PKS 中,trans‑AT PKS 的KS 也能形成与其底物的化学结构紧密相关的进化分支[53]。Jorn Piel课题组[54]利用KS数据库检索发现了与mis PKS(misakinolides PKS)进化上非常接近的序列,最终鉴定其产物为二聚的大环内酯luminaolide B(6,结构见图5)。通过研究misakinolides、scytophycin 以及 luminaolides 的生物合成和进化关系,发现它们的基因簇来源于共同祖先,通过缺失或获得PKS上游或末端序列实现结构多样化。为了实现trans‑AT PKS 产物的结构预测,理解trans‑AT PKS 的生物合成基础和进化模式,Jorn Piel 和Marnix Medema 课题组[55]开发了在线工具transATor 和transPACT。TransATor 输入PKS 序列,预测KS 底物特异性及对应的聚酮核心结构,利用这一工具他们发现了tartrolon 类化合物和leptolyngbyalide。TransPACT是trans‑AT PKS 注释和比较工具,可以自动形成KS 的功能分支,识别不同PKS 组装链共有的连续模块。他们利用transPACT 从GenBank 中得到1782个trans‑AT PKS的基因簇并用antiSMASH进行分析,随后提取KS 序列进行进化分析,根据计算生成的模块共享网络和进化树进行基因挖掘,最终发现了新的trans‑AT PKS 产物secimide(7)、gynuellalide(8)、spliceostatin L(9)等(结构见图5),并探究了相似化学结构在序列上的相关性[56]。这些研究表明了可以通过对trans‑AT PKS中KS 结构域的进化分析来指导结构新颖的聚酮化合物的挖掘,同时也为PKS 改造以生产非天然的trans‑AT PKS 聚酮产物提供了基础。辛志宏课题组[57]利用KS 序列的进化分析挖掘植物内生真菌中Ⅰ型PKS 产物,发现了具有抗菌活性的天然色素talafun(10,结构见图5)。该研究表明使用高度保守的KS 结构域作为进化标记,可以快速连接真菌基因信息和化学结构,并作为高通量测序技术的常规方法应用于实践。
除了PKS 本身,同一个簇中的共同进化基因同样可以用于基因挖掘。烯二炔是一类由Ⅰ型PKS 产生的线性多烯,具有极高活性,常作为临床试验中的抗体药物配合物[58]。Shen Ben 课题组[58‑59]以两组不同的烯二炔生物合成基因E5/E 和E/E10 为靶标通过实时定量PCR 从3400 株菌株中挖掘烯二炔化合物。通过PCR 他们发现了81 株具有烯二炔聚酮合酶基因的菌,同时对基因E的进化分析表明许多簇与已知的是不同的。为确认这一结果,他们对31株代表性菌株进行了基因组测序,对其中的相关基因簇进行GNN(genome neighborhood network)分析,发现了与已知基因簇明显不同的基因簇,最终通过分离鉴定发现了活性化合物tiancimycin A(11,结构见图5)[58‑59]。以上研究为挖掘更多烯二炔类化合物或利用PKS 合成烯二炔同系物奠定了一定基础。
2.4 PKS生物合成改造
事实上,自从三十多年前Ⅰ型PKS 的多模块化特征被确定,研究人员就尝试利用模块和结构域的重新组合来产生新的非天然聚酮化合物[60]。由于其多模块的特性,PKS 提供了一个通用的合成平台,例如可以作为合成特定有机酸的有效方法[61]。然而,在对PKS 装配线进行改造的一些早期尝试中,交换或删减一些结构域和模块经常导致酶活性显著降低甚至是无活性[52],推测这与蛋白相互作用[53]以及底物选择性[54]有关。
同时,越来越多的证据表明,对组装线系统的进化有更好的理解可以进一步提高改造这些系统的能力[图6(a)][29,35,62]。Adrian Keatinge‑Clay课题组[63‑65]根据新定义的模块边界,即在KS 和AT 之间选择切点构建了多个杂合PKS,目标产物的产量得到明显的增高(相比于根据传统定义构建的杂合PKS 有10~48 倍的提高)[图6(b)]。Christian Hertweck 课题组[66]分析几条聚酮合成基因簇中各个模块KS 的系统发育树,通过几种不同方式的剪切融合,证明在自然进化过程中模块的增加或删减可能发生在KS‑AT 连接处,并通过分析基因簇中P450 修饰酶的底物特异性,进一步推测出PKS 的进化顺序。同样地,除了上述KS‑AT连接处,post‑AT 连接处同样被证明为有效的模块融合和结构域交换切点[67‑69]。类似的利用自然偏好的位点进行“剪切‑粘贴”的策略可能也适用于trans‑AT PKS的改造[54]。
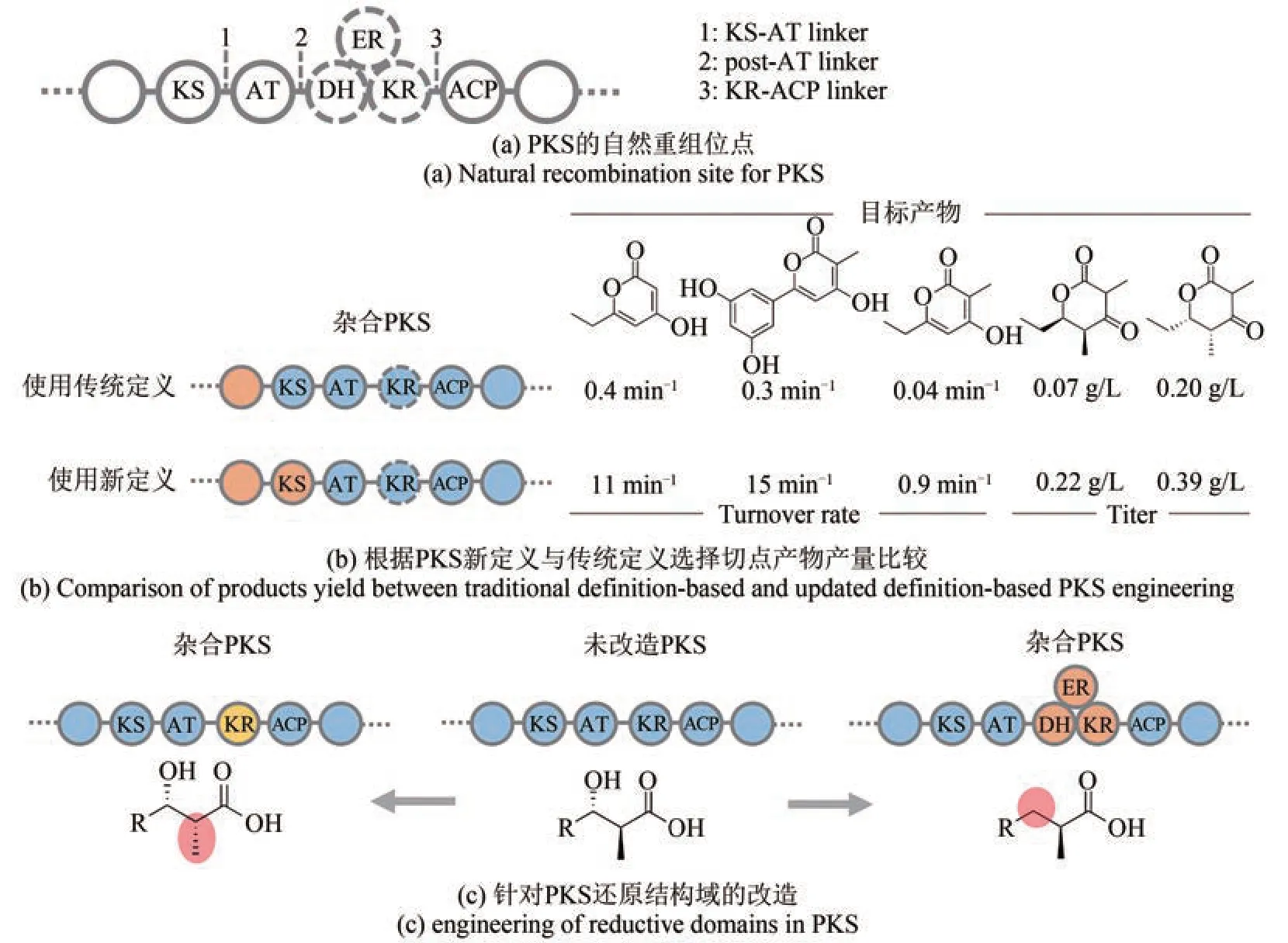
图6 进化导向的PKS改造Fig. 6 Evolution‑guided engineering for PKS
Jay Keasling课题组[69‑72]通过序列比对,把KR或KR‑DH‑ER 整体在模块间进行交换,体外和体内实验证明了这种策略的可行性[图6(c)],这也进一步表明还原结构域可能为进化过程中潜在的重组单元。另一方面,一些研究通过尝试对AT[73]和KR[74]活性口袋进行多点突变以逆转其选择性,然而当这些突变被用于整个模块,仍无法专一地得到预期产物,可以推测PKS 并不是单纯依赖点突变的方式,而是通过基因重组导致的结构域交换去改变结构域的选择性[29]。
虽然PKS 单个或多个结构域甚至整个模块的结构已经通过X 射线单晶衍射和低温冷冻电镜技术解析[75‑81],揭示了蛋白质相互作用在聚酮链延伸各个阶段的重要性以及一些重要的蛋白相互作用位点,但PKS 催化过程中各个结构域潜在的协同作用未被完全揭示,基于三维结构的理性设计改造仍困难重重。因此,从自然进化的角度分析整个PKS 或各个结构域的进化关系,推测自然重组发生的位点,以此作为人工改造的切点,并根据进化关系选择合适的候选PKS 进行拼接构建杂合PKS,这为研究者提供了进化导向的PKS 改造新思路。
3 进化和大数据导向的NRPS研究
非核糖体肽合成酶(NRPS)是来自细菌和真菌的多模块酶或酶复合体,其催化生成的肽类化合物很多具有重要的生物活性,其中一些被临床使用如环孢素、万古霉素、达托霉素等[82‑84]。根据合成酶整体结构的不同,NRPS 通常被分为Ⅰ型和Ⅱ型[85]。Ⅰ型NRPS 是大型模块化复合体,以类似于Ⅰ型PKS的流水线方式生成肽类化合物,每个模块主要包含C(condensation)、A(adenylation)、T(thiolation,也称载体蛋白)3 个结构域或其他修饰型结构域如E(epimerization)。Ⅱ型NRPS 蛋白通常是独立的酶或两个结构域协同形成独特的氨基酸衍生物[85]。NRPS 合成肽类化合物过程中,A结构域选择特定的氨基酸单体,由ATP激活形成氨酰基‑AMP,然后转移到载体蛋白T 上。C 结构域缩合被激活的氨酰基(肽基)硫酯,通过形成酰胺键进行链延伸。同PKS 一样,NRPS 对挖掘活性分子、研究酶催化和蛋白相互作用等具有重要意义。
3.1 NRPS进化机理
与PKS 类似,NRPS 的进化过程中自然发生的基因重组发挥了重要作用。非核糖体肽的分化主要由A 结构域或子结构域的重组驱动[86]。A 结构域内的重组发生在Acore的可变部分从而调节底物,但结构域之间的相互作用以及Asub基本不受影响[62,87]。
NRPS 的另一核心结构域是C 结构域,根据立体选择性的不同可以分为LCL、DCL和起始C 结构域(CS或starterC)3种类型。尽管LCL和起始C 结构域由于某些序列的显著差异导致了底物的差异(氨基和β‑羟基羧酸),它们在进化树上的关系似乎比其他亚型更密切[88]。已有研究表明C 结构域对立体化学的选择与E 结构域功能相关[89]。在细菌NRPS中PCPE‑E‑DCL几乎是普遍保守的,表明尽管在进化史上发生了无数的基因组复制、插入、删除和重组事件,E‑DCL连接域仍保持着强大的选择压力[89]。
3.2 NRPS功能预测
根据NRPS 的进化机理,NRPS 组装线上的结构域序列与产物的化学结构之间有直接的关系,这种关系使从DNA 序列预测肽类化合物的化学结构成为可能。
1991年,Stachelhaus和Marahiel报道了A 结构域底物特异性预测的开创性工作。他们关注到A结构域的系统进化树与底物类别有较强的关联性(图7),其次发现在Acore区域形成底物结合口袋的10 个关键氨基酸序列(即Stachelhaus 密码),与它们接收的底物具有高度相关性[90]。随后,一些NRPS A 结构域的底物预测工具被开发利用,如NRPSpredictor2[91]和SANDPUMA[92]等。这些预测工具的发展一方面为基因挖掘发现新型天然产物提供巨大帮助,另一方面也可为NRPS装配线改造寻找合适的候选基因。

图7 进化导向方法预测A结构域底物示意图Fig. 7 Prediction for substrates of A domain using phylogeny‑guided method
NRPS 的 A 结构域相当于“底物水平”的进化信号,可以用来预测底物特异性,而NRPS 的C 结构域相当于“途径水平”的进化信号,可以用来预测类似分子的BGC 模式[93]。C 结构域超家族除了原始C 结构域,还包括其他几个同样属于NRPS结构域的成员如CS、DCL、LCL、E(差向异构化)、Cyc(杂环化)、DualC(差向异构化/缩合)和modAAC(脱氢氨基酸相关),它们由于功能不同在进化树上形成了明显分支(图8)[89,94],因此可以利用C结构域的进化分析进行NRPS中相关结构域的功能预测。
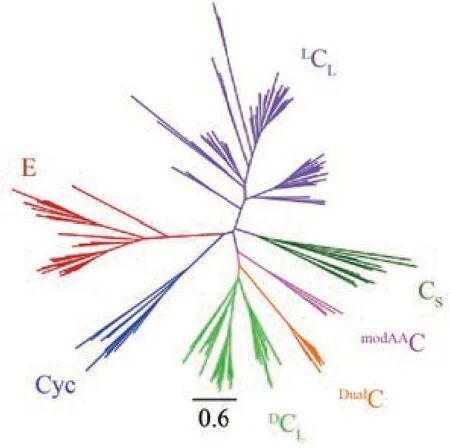
图8 C结构域超家族的无根进化树[89]Fig. 8 Unrooted phylogenetic tree of the C‑domainsuperfamily [89]
3.3 NRPS基因挖掘
NRPS 中A 结构域和C 结构域的底物选择性以及不同模块的催化顺序决定了氨基酸的连接顺序,意味着非核糖体肽类化合物的结构可以与NRPS序列直接关联起来,因此可以利用 A 结构域或C 结构域的进化分析进行NRPS的基因挖掘。
钙离子依赖型抗生素是一类需要钙离子才能发挥活性的环肽。已知的这类化合物都有保守的Asp4‑X‑Asp6‑Gly7片段促进其与钙离子的结合。基于此,Sean Brady 课题组[95]通过PCR 从土壤eDNA 中扩增NRPS 的A 结构域,并对扩增得到的序列用eSNaPD 进行分析。标签序列的进化树中Asp4结构域有许多进化分支与已知BGC 距离较远,这暗示土壤微生物组中有未知的钙离子依赖抗生素存在。通过异源表达,他们鉴定出一类新的钙离子依赖抗生素malacidins(12,结构见图9)。虽然基于宏基因组的抗生素发现方法仍处于起步阶段,但是上述研究中规模化和自动化方法为高效挖掘隐藏在宏基因组中的抗生素,以及为对抗抗生素耐药性提供了一种潜在的强大方法。
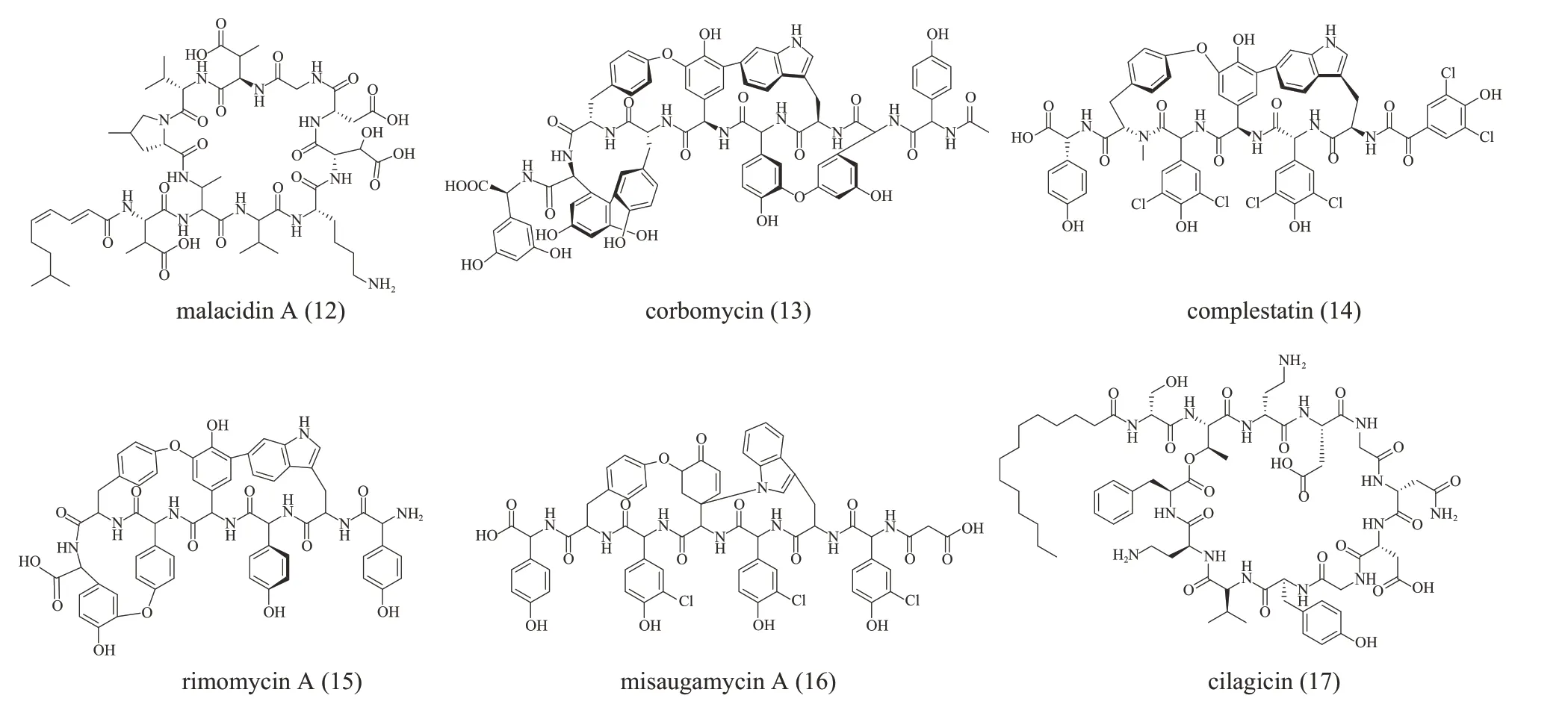
图9 进化导向基因挖掘获得的肽类化合物12~17分子结构(化合物12为基于NRPS的A 结构域基因挖掘发现的钙离子依赖抗生素;化合物13~16为基于NRPS的C结构域基因挖掘发现的糖肽类抗生素;化合物17为基于NRPS预测发现的脂肽类化合物)Fig. 9 The structure of peptide molecules 12~17 obtained by phylogeny‑guided genome mining(Compounds 12 was calcium‑dependent antibiotic discovered by genome mining of A domain; compounds 13~16 belonged to glycopeptide family of antibiotics discovered by genome mining of C domain;compound 17 was lipopeptide antibiotic discovered by NRPS prediction)
2020 年,Gerard Wright课题组[96]收集了71个糖肽类抗生素的基因簇,利用基因簇中的C结构域构建进化树预测出可能有新的生物活性的糖肽类化合物,最终发现两个具有新的功能的糖肽类抗生素corbomycin(13)和complestatin(14)(结构见图9),并阐明它们是通过与肽聚糖结合,阻断了自溶素(自溶素是生长过程中细胞壁重构所必需的肽聚糖水解酶)作用从而抑制细菌生长。近期,Gerard Wright 课题组[97]利用糖肽抗生素指纹序列扩充候选BGC 后,通过进化导向基因挖掘及异源表达鉴定了5 个新的typeⅤ糖肽类抗生素(typeⅤGPA)rimomycins(15)和misaugamycins(16)(结构见图9),并证明它们的作用机理也是通过阻止自溶素活性而抑制细胞分裂。这些发现拓展了typeⅤGPA的化学多样性,为药物开发提供了新的化学骨架,展示了基于进化的生物信息学平台在挖掘糖肽类抗生素化学“暗物质”中的巨大应用潜力。
NRPS 的准确预测也促进了不依赖于传统分离技术的生物活性肽的开发。Sean Brady 课题组[98]利用对脂肽的生物信息学预测,通过化学合成获得了具有较强抗菌活性的脂肽cilagicin(17,结构见图9),并阐明它是通过阻断细胞壁生物合成中两种必需的十一烯基磷酸发挥抗菌作用。该研究基于CS结构域的进化树分析,发现了孤儿BGC,然后将化合物结构预测与化学合成结合得到对应产物,避免了目标基因簇不表达或产物产量低等问题。
3.4 NRPS及NRPS‑PKS杂合的生物合成改造
3.4.1 NRPS
目前针对NRPS 的生物合成改造已有不少尝试,主要可以分为以下几类:①取代A 结构域或A‑T 结构域,从而改变延伸单元[99];②改变A 结构域的底物结合口袋[100‑101];③C‑A 或者C‑A‑T 结构域交换[102]。
通过A 结构域取代,David Ackerley 课题组[86]高效地获得了高产量的pyoverdine修饰肽,确定了允许的A 结构域重组边界[图10(a)]。Jorn Piel课题组[103]对hormaomycin 合成基因簇中7 个A 结构域核苷酸序列分析发现,除了与底物识别口袋有关的约400 个碱基对,其余序列展现出了超过90%的相似度,这暗示着潜在的天然重组位点。基于这种猜测,利用从自然重组推断的序列边界,以HrmO 第3 个A 结构域作为模板构建了3 个嵌合体,体外试验表明A 结构域的底物特异性被成功转移,并且仍保持较高的转化率。基于同样的策略,Donald Hilvert 课题组[104]把9 个不同的底物特异性移植到gramicidin S 合成基因簇的GrsA 模块中[图10(b)]。

图10 进化导向的NRPS改造Fig. 10 Evolution‑guided engineering for NRPS
C 结构域和C‑A 连接域影响A 结构域催化活性和底物选择性。与此类只改变A 结构域核心序列的策略不同,Helge Bode 课题组[105‑108]将A‑T‑C或A‑T‑C/E 定义为交换单元,提出了XU(exchange unit)以及XUC(exchange unit condensation domain)的概念。他们认为C‑A 连接域是理想的重组位点,因为序列比对表明A‑T 和T‑C 连接域保守性低,并且在催化循环中可能参与重要的蛋白相互作用,而C与A结构域之间主要依赖疏水作用。依据这种交换单元构建的杂合NRPS 虽然相比于野生型显示出降低的产量,但仍可以得到足够用于活性分析的目标产物[图10(c)],且在少数例子中实现了与野生型同等的产量。有趣的是,这种交换单元类似于PKS 模块边界的新定义,考虑到C 结构域同样具有门控的功能,可以推测C 结构域与上游A‑T结构域进化关系上可能具有一定的关联性。
3.4.2 NRPS/PKS 杂合
聚酮和多肽类化合物具有截然不同的骨架,而NRPS和PKS杂合大大促进了天然产物类型的多样化。对NRPS/PKS组装线进行改造是产生新型生物活性分子的一种有效方法。在真菌中,高度还原型聚酮合酶(HR‑PKS)可以与NRPS形成杂合,合成以吡啶酮骨架为代表的一系列真菌聚酮化合物。Hideaki Oikawa 课题组[109]分析了NCBI 真菌基因组中884 条PKS‑NRPS 杂合酶,发现了酶系统进化树的分支与产物分子骨架之间有明显的对应关系,为真菌PKS‑NRPS基因簇产物的分布与结构多样性提供了宏观见解。
细菌PKS‑NRPS杂合酶是合成博来霉素等的模块型酶。尽管PKS和NRPS的进化机制仍存在许多疑点[29],但通过进化分析发现一些自然重组位点,可以作为模块改造和结构域融合的切点。双内酯缩肽类化合物antimycins 由NRPS‑PKS 杂合酶合成,序列分析表明其可能与三内酯JBIR‑06、四内酯neoantimycin A 由同一个祖先进化而来。Ikuro Abe 课题组[110]受此启发,进一步推测出自然重组发生的位点,对JBIR‑06 和neoantimycin A 合成酶模块进行增减,通过异源表达实现了对内酯缩肽类化合物环尺寸大小的控制。这项研究通过分析NRPS‑PKS杂合酶自然重组进化的过程,证实上述PKS 和NRPS 改造策略(2.4 和3.4.1 小节)同样适用于NRPS‑PKS杂合酶体系[110]。
4 进化和大数据导向的非模块型酶研究
4.1 非模块型生物合成酶基因挖掘
4.1.1 RiPP (ribosomally‑synthesized and post‑translationally modified peptide)合成酶
不同于聚酮、非核糖体肽类化合物等,RiPP的生物合成途径缺乏共同的生物合成特征,其基因簇难以进行可靠的生物信息学预测[111]。针对RiPP 的预测工具可以依赖于前体肽特征或者修饰酶。丁伟课题组[112]通过深度学习来探究RiPP 生物合成的底层逻辑,提出了基于BERT 预训练模型的组合模型BERiPP(bidirectional language model for enhancing the performance of identification of RiPP precursor peptide)。BERiPP 能够在不考虑基因组背景的情况下无差别地识别RiPP 前体肽,并对前导肽裂解位点进行预测,为高通量挖掘新的RiPP提供了思路。
此外,基于机器学习技术的进步,Nathan Magarvey 课题组[115]开发了DeepRiPP,集成了基因组和代谢组学数据,使用机器学习来自动发现和分离新的RiPP。DeepRiPP 通过3 个模块实现:识别独立于基因组结构和邻近生物合成基因的RiPP,优先选择编码新化合物的基因座,从复杂的细菌提取物中自动分离出相应的产物。他们利用DeepRiPP 对来自463 株菌株的10 498 个提取物的数据库进行大规模比较代谢组学分析,最终发现了3 种新型RiPP,结构与平台预测的完全一致。DeepRiPP 提高了RiPP 基因挖掘效率,展示了机器学习技术在微生物基因大数据挖掘中的应用前景。
4.1.2 萜类合酶
萜类化合物是真菌及植物中常见的重要天然产物类型,由IPP(isopentenyl diphosphate)和DMAPP(dimethylallyl diphosphate)为底物形成单萜、倍半萜和二萜等生物合成的线性前体,再由萜类合酶催化多样的环化反应形成复杂的碳骨架结构。与植物和真菌萜类合成酶相比,细菌萜类合酶总体上序列相似度很低。Dickschat 和Garbeva课题组[116]对链霉菌进行了全基因组系统发育分析,比较了萜类合酶基因在链霉菌中的分布,并对这些萜类合酶进行进化分析,研究发现这些酶的进化与链霉菌的进化并不一致,这表明基因水平转移可能是链霉菌萜类合酶基因分布的重要机制。同时,他们发现链霉菌的萜类合酶在进化树上可分为10 类,其中土臭素(geosmin)合酶最为丰富。为探究细菌萜类合酶和真菌萜类合酶的进化关系,Chen Feng 课题组[117]对908 个真菌萜类合酶和1535 个细菌萜类合酶进行进化分析,研究表明真菌同样通过基因水平转移从细菌中获得萜类合酶。此外,近年来越来越多的证据表明基因水平转移在萜类合成前体生物合成途径的进化中也起着重要作用[118]。
二萜是由4 个异戊二烯单位构成的萜类化合物,广泛分布于植物界,其含氧衍生物很多具有较强的生物活性,如紫杉醇、雷公藤内酯等。微生物代谢产物中也发现有二萜类化合物,但与植物来源二萜合酶相比,来自菌类的二萜合酶的研究较少。为了从公共数据库中挖掘潜在的二萜合酶编码序列,刘宏伟课题组[119]利用EriG蛋白(猴头菇中形成cyathane 骨架的环化酶,属于UbiA 超家族)序列作为探针进行基因组挖掘,通过序列聚类分析和进化树分析,发现了细菌和真菌中与UbiA 相关的新家族二萜环化酶(cluster 11)。通过在大肠杆菌中表达鉴定了7个新的二萜环化酶,并确定了其对应产物的结构,其中包括一个新的具有不同寻常骨架的二萜lydicene。这项研究丰富了细菌和真菌中二萜环化酶的多样性,更新了UbiA超家族成员,为微生物二萜合酶在生物催化和代谢工程中的应用提供了新的机遇。
近期,Dickschat和Abe课题组[120]开展了真菌二倍半萜生物合成酶的系统进化研究。二倍半萜是由C 端异戊烯转移酶(prenyltransferase,PT)和N 端Ⅰ型萜类合酶两个结构域组成(terpene synthase, TS)的嵌合萜类合酶(PTTS)催化合成,该课题组利用18 个PTTS 的TS 功能区域构建系统进化树,发现PTTS 形成6 类主要分支,对应不同的环化产物[120]。刘天罡课题组[120]进一步拓展了PTTS 进化树分析,结合基因挖掘揭示了6 类主要分支大致对应异戊二烯线性前体的两大环化模式,即第四‑第五双键成环的Type A 反应以及第三‑第四双键成环的Type B 反应。利用PTTS 进化树的基因挖掘,刘天罡课题组[121]首次发现了三种真菌来源三萜合酶以及其三萜产物。其中三萜合酶MpMS 和CgCS 催化的环化模式是不符合上述两类的新机制,表明基于序列分析的萜类合酶功能预测的局限以及其丰富的催化可塑性。
4.1.3 其他基因
在PKS 中,催化Claisen 缩合反应形成聚酮主链的KS 属于硫解酶超家族[122]。从进化角度来说,这个超家族的成员具有与分支相同的功能簇,都从类似于古菌硫解酶的类硫解酶祖先分化而来。考虑到它们进化的多样性和结构的相似性,Ramon Gonzalez课题组[123]假设硫解酶超家族中除了PKS 以外还有酶能够催化迭代Claisen 缩合反应合成聚酮骨架。他们通过合成具有代表性的聚酮化合物,如内酯(三乙酸内酯)、烷基间苯二酚酸(alkylresorcinolic acids)、烷基间苯二酚、羟基苯甲酸和烷基酚等证明了这一途径的可行性。这一发现可以扩展到其他硫解酶,以进一步阐明它们的结构和功能关系,并将它们的生物合成潜力用于PKS研究[123]。
末端炔是一种广泛应用于有机合成、医药科学、材料科学和生物化学的功能性物质,在微生物体内可以由特殊的去饱和酶--乙炔酶催化形成[124]。Zhang Wenjun 课题组[125‑126]阐明了JamA、JamB、JamC 在末端炔jamaicamide 生物合成中的功能,以炔基形成关键酶jamB基因作为探针对其序列相似基因进行进化分析,筛选新的炔烃基因簇,发现了由TtuA、TtuB 和TtuC 组成的新的末端炔生物合成机制,从而扩大了末端炔烃生物合成研究模型,在合成和化学生物学中有广泛的应用前景。
吲哚咔唑是一种被用作抗癌药物先导的天然产物, 其核心结构是由嗜铬吡咯酸(chromopyrrolic acid, CPA)合酶催化的两分子氧化的色氨酸二聚形成。对土壤宏基因组中的CPA 合酶同源基因进行系统发育分析,发现了新的吲哚色氨酸boregomycins A~D、erdasporine A~B 以及reductasporine 等[15,127‑128]。
安莎霉素是临床上重要的天然产物家族,这类化合物的显著结构特征是存在一个芳香核心,来自共同的前体3‑氨基‑5‑羟基苯甲酸(AHBA)。对链霉菌AHBA 合酶同源基因进行进化分析,最终从6株菌中发现了25个安莎霉素,包括8个新的juanlimycin和neoansamycin等化合物[15]。
进化导向基因挖掘方法对化合物生物合成特征基因如催化末端炔形成的乙炔酶、催化吲哚咔唑形成的嗜铬吡咯酸合酶以及催化安莎霉素形成的AHBA 合酶进行进化分析,从中挖掘出具有相同官能团的活性化合物,该方法使得利用antiSMASH 或ClusterFinder[129]无法直接检测到的特殊化合物BGC 的检索成为可能,为发展基于进化的生物信息学途径提供了可行性验证。
4.2 靶向抗性基因的天然产物挖掘
天然产物挖掘的主要目标之一就是发现新的作用模式的抗生素,以抵抗病原菌的多重耐药性。为避免产生的抗生素对自身的伤害,微生物进化出了几种避免自身毒性的耐药策略,包括产物修饰、底物运输和结合、靶标复制或修饰,这些耐药修饰由位于抗生素BGC 附近的抗性基因编码[130]。基因簇中抗性基因的存在可以作为预测该途径合成的天然产物生物活性的窗口。基于自我抵抗基因识别的天然产物发现有助于弥补活性导向和基因组导向方法在天然产物发现和功能分配中的缺口[10,131]。近年来,也有一些利用进化思想靶向抗性基因进行的天然产物挖掘,例如前文中提到的ARTS即是靶向抗性基因的进化导向基因挖掘工具[16‑17]。 利用ARTS 探测模式, Nadine Ziemert 课题组分析了所有已知的BGC 和可用的细菌基因组(其中包含已知的耐药靶基因),除了MIBiG 数据库中26 个已知的基因簇,还检测到22个具有抗性靶标的基因簇,体现了进化导向基因挖掘的应用潜力。
转录调控因子tetR/marR和抗性转运因子如tetA是四环素生物合成中常见的一对抗性基因,其中抗性转运蛋白是位于细胞膜上的四环素/金属质子逆向转运蛋白,而调控蛋白TetR 是四环素诱导阻遏蛋白。基于四环素BGC 的抗性机制,戈惠明课题组[52]以TetR/MarR‑转运蛋白作为挖掘四环素类天然产物的指标,结合链长因子CLF 的系统发育分析进一步细化,发现了25 个不同的四环素基因簇,最终分离出一种新的四环素海南环素。这种同时靶向抗性基因和Ⅱ型PKS 基因的基因挖掘方法为特异性、高效地挖掘新颖的抗生素提供了可能。
4.3 进化导向的非模块型酶改造
基于进化分析所启发的生物合成酶改造并不局限于PKS 和NRPS 这类多模块化酶。Tobias Erb课题组[132]通过将烯酰辅酶A 羧化/还原酶(ECR)与其他中链脱氢/还原酶(MDR)进行聚类分析(图11),发现丙酰辅酶A合酶(PCS)以及古菌烯酰还原酶(AER)可能与ECR 由共同的祖先进化而来,并具有潜在的CO2结合口袋和羧化功能。在一定CO2浓度下,PCS 和AER 均展现出较弱的羧化功能,但主要是还原产物(大于95%)。结合三维结构模型,作者进一步分析了CO2结合口袋的序列,并对关键氨基酸进行突变。通过增强CO2的结合和阻止水进入口袋,成功唤醒了PCS 和AER 的羧化功能,羧化产物占比均提高了约20 倍,成为主要产物(图12)。

图11 中链脱氢/还原酶(MDR)进化分析Fig. 11 Phylogenetic analysis of MDR
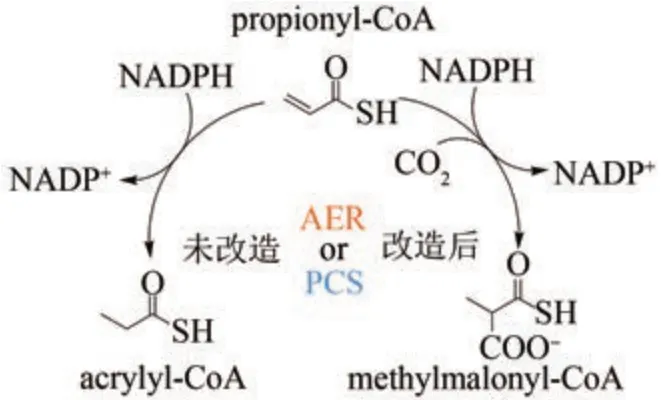
图12 古菌烯酰还原酶(AER)和丙酰辅酶A合酶(PCS)的改造Fig. 12 Engineering for AER and PCS
上述的研究是通过对同源蛋白的比较来阐明特定蛋白功能的进化,这种方法通常称为“水平的”。因为它们是基于对某一进化阶段的分析,用于分析的蛋白都是在现存物种中发现的蛋白质。而系统发育算法为序列分析增加了垂直维度,可以根据现存序列的进化追溯到共同的祖先[133]。祖先序列重建(ancestral sequence reconstruction,ASR)即是实现从现代序列(即现存序列)推断原始序列的强大工具[134]。ASR 的一个基本元素是系统发育树的计算,其叶子是所选的现存序列,与系统发育树的根相关的重构序列代表了所研究序列的共同祖先。如果这个序列编码了一种蛋白质,就可以通过基因合成技术“复活”这个祖先蛋白,并借助生物化学实验来研究其生化特性。ASR 还可以推导出树中所有内部节点的序列,进一步阐明进化过程[133]。
5 总结与展望
BGC 可以通过广泛的水平基因转移有效地分散,甚至跨越门的边界,因此天然产物研究中基于进化的发现策略在增加新颖性方面有力地补充了传统方法。基于进化原理的生物信息学分析工具的开发和应用产生了日益增长的遗传(基因)、催化(蛋白)和化学(化合物结构)数据库,并推动天然产物研究进入现代大数据时代,使得天然产物全景图可视化成为可能[1‑2,6]。通过这些策略获得的天然产物不仅加深了对天然生物活性分子合成方式的认识,而且丰富了生物活性化合物库。
随着天然产物相关研究的数量和质量的增长,人工智能分析方法(如机器学习)的应用潜力也在增加,将进化导向的方法与人工智能结合将是该领域的发展方向之一。而成功的机器学习方法需要高质量的训练数据,未来可能需要跨实验室甚至国际的协调努力,以标准化的方式生成数据集并进行管理[135],而这依赖于生物信息学的发展[136]。
基于进化及大数据进行的天然产物挖掘不仅本质上受到已测序基因组数据量与范围的限制,目前还仍然面临一些挑战:①许多含有目标分子BGC 的微生物在实验室条件下不可培养,或者目标基因簇不表达。目前对于这一问题的解决,主要是依赖于异源表达等分子生物学技术的发展,因此,提高异源表达效率等技术方面的改进将促进进化导向的天然产物挖掘。②预测基因簇产物的生物活性仍然困难。目前只能通过靶向生物活性分子类似物或者抗性基因的天然产物挖掘提高发现活性分子的概率。探寻化合物结构与生物活性在进化中的关系或许能够为进化导向天然产物挖掘发现新的活性分子提供更多机会。③萜类、生物碱等的BGC 很难展现化合物的结构特征,基因簇产物的分子结构预测仍有较大挑战。对于这些非模块型BGC,还需要增加基因簇与其产物的表征数量,并仔细分析每一步生物合成酶的进化特征和规律。
酶改造方面,对于模块型酶的合理改造是自其发现以来的重要目标。在PKS及NRPS改造的早期尝试中,交换或删减一些结构域和模块经常导致酶活性显著降低甚至是无活性,而对组装线系统的进化分析推断出自然重组发生的位点,可以指导人工酶改造的设计。目前,基于进化分析得出的PKS 新模块定义以及NRPS 的XUC 概念为模块型酶的改造提供了理论依据,这些概念的应用以及进一步优化将推动合成生物学的发展。此外,这种思路不局限于PKS和NRPS这类多结构域、多模块的酶,将进化与大数据分析相结合也将为其他酶的改造提供新的思路。
自然界千万年来基于各种进化机制创造了丰富多样的生物合成途径和天然产物,人类认识自然界的脚步也从未停歇。通过生物信息学研究生物合成酶进化的机制,挖掘其活性产物用于医药健康或者农业生产领域,结合大数据分析描绘天然产物全景图,或者利用自然界的规则和元件,从模仿进化的角度去设计改造生物合成酶以满足人类的需求,这正是发现自然并改造自然的过程。
