CRISPR/Cas 基因组编辑技术在丝状真菌次级代谢产物合成中的应用
2023-09-16林继聪邹根刘宏民魏勇军
林继聪,邹根,刘宏民,魏勇军
(1 郑州大学药学院,郑州大学药物研究院,郑州大学合成生物学实验室,药物关键制备技术教育部重点实验室,河南郑州 450001; 2 上海市农业科学院食用菌研究所,农业农村部南方食用菌资源利用重点实验室,国家食用菌工程技术研究中心,上海 201403)

丝状真菌(filamentous fungi)能够合成抗生素、色素、酶、激素等多种产品[1],在医药、化工、农业和基础生物学研究中广泛应用。黄曲霉(Aspergillus flavus)会分泌对人类或动物具有致病性的黄曲霉毒素[2];产黄青霉(Penicilliumchrysogenum)、顶头孢霉(Acremonium chrysogenum)和土曲霉(Aspergillus terreus)能合成β‑内酰胺类抗生素[3‑4](青霉素和头孢菌素)或他汀类药物[5](图1)。黑曲霉(Aspergillus niger)是葡糖淀粉酶[6]和果胶酶[7]的主要生产宿主,里氏木霉(Trichoderma reesei)是纤维素酶的主要生产宿主[8];丝状真菌生产的酶占据工业酶市场份额的50%[9]。部分丝状真菌的次级代谢产物在特定环境条件下产生,但难以在实验室的生长条件下合成[10]。丝状真菌中含有大量“沉默的”生物合成基因簇(biosynthetic gene clusters,BGC),这些合成基因簇具有合成活性天然产物的潜力[11]。
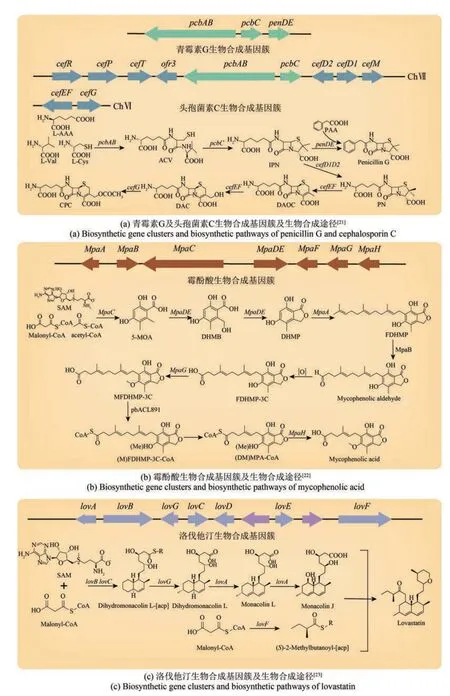
图1 部分次级代谢产物生物合成基因簇及生物合成途径[21‑23]Fig. 1 Biosynthetic gene clusters and biosynthetic pathways of some secondary metabolites
功能基因组研究对真菌次级代谢产物的合成具有重大影响[12]。基因组研究需要对宿主基因组进行缺失、插入和替换等遗传操作,以探究基因的功能及其与其他基因的相互作用。与细菌和酵母相比,丝状真菌的遗传背景较为复杂[13];在进行分子水平研究时,可用的遗传筛选标记有限。此外,丝状真菌的同源重组效率低,生长缓慢,遗传操作困难,阻碍了对其的工程改造[14],限制了对其的进一步开发应用[15]。
基因组编辑技术为探究真菌生理特性、合成多种天然产物等提供了技术基础,也是获取高性能工业生产菌株的关键。目前,基因组编辑技术主要有锌指核酸酶技术(zinc‑finger nucleases,ZFN)、类转录激活因子效应物核酸酶(transcription activator‑like effector nuclease proteins,TALEN)以及成簇的规律间隔短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)[16]等三种。其中,CRISPR 系统最简便,在丝状真菌遗传育种及基因改造方面得到广泛应用[17‑18]。本文对丝状真菌次级代谢产物(secondary metabolites,SM)合成以及基因组编辑技术进行了综述,并阐述了丝状真菌CRISPR 介导的基因组编辑技术的发展及其在次级代谢产物合成等方面的应用趋势。
1 丝状真菌次级代谢产物
丝状真菌次级代谢产物是药物的重要来源之一。青霉素类、头孢菌素类药物、灰黄霉素[19]等均属于丝状真菌次级代谢产物[20](图1)。
弯颈霉属(Tolypocladium)的环孢菌素被用来预防器官或组织移植产生的排斥反应[24]。短密青霉(Penicillium brevicompactum)的霉酚酸可以抑制免疫排异反应,在医药领域具有重要的应用价值[25]。高血脂会增加血液的黏稠度,且可能引发动脉粥样硬化、高血压、冠心病等严重疾病。土曲霉(Aspergillus terreus)和橘青霉(Penicillium citrinum)的他汀类活性天然产物是降血脂的首选[26]。洛伐他汀是一种重要的降脂药,但与其相比,辛伐他汀的降血脂作用更强,是效果更好的降血脂药物。在工业上,先运用土曲霉合成洛伐他汀,然后通过碱水解的方法获得了中间产物莫纳克林J,再通过化学方法合成辛伐他汀。吕雪峰团队[27]鉴定了一个新的水解酶PcEST,该酶可以高效转化洛伐他汀为莫纳克林J。同时,研究人员利用土曲霉内源性组成型强启动子PgpdAt过表达特异性转录调控因子lovE,将莫纳克林J的产量提高了52.5%[28]。
丝状真菌次级代谢产物的合成过程复杂,相关基因通常成簇排列(图1)。AntiSMASH等生物信息学软件可以预测基因组中的BGC,但仍需通过敲除等基因操作研究证实相关BGC的功能[29]。天然宿主在实验室条件下生长缓慢且遗传工具稀缺,部分天然产物BGC难以表达;尽管可在模式宿主中异源表达天然产物BGC,但成功率较低[30]。通常,酿酒酵母(Saccharomyces cerevisiae)是异源合成天然产物BGC的真菌宿主[31]。酿酒酵母的高效重组方法和多种基因组编辑技术为复杂天然产物合成途径的异源表达提供了基础。青蒿酸[32]、人参皂苷Rh2[33]、文多灵和长春质碱[34]等多种复杂植物天然产物已在酵母中成功合成,但长片段天然产物BGC的表达仍较为困难。开发真菌类基因组编辑系统是深入研究和表征天然产物BGC的重要途径。
2 CRISPR/Cas基因组编辑技术
2013 年,新一代基因编辑技术CRISPR/Cas9诞生[35],开启了基因组编辑的新时代。CRISPR/Cas9 系统作为细菌的一种特殊防御机制,主要用来抵御入侵的病毒和质粒[36]。DiCarlo 等[37]在酿酒酵母中实现了基于CRISPR/Cas9 的基因编辑。2015 年,Liu 等[38]第一次将CRISPR/Cas9 技术应用于里氏木霉,为CRISPR/Cas9 基因组编辑系统广泛应用于丝状真菌的遗传改造奠定了基础。与ZFN 和TALEN 技术相比,CRISPR/Cas9 系统能够诱导宿主细胞中DNA 的双链断裂(double strand break,DSB),提高了基因编辑整合的效率。
2.1 CRISPR/Cas9系统的工作原理
CRISPR/Cas9 基因组编辑系统由Cas9 核酸酶以及通过融合CRISPR RNA(crRNA)和trans‑activating crRNA (tracrRNA) 形成的 sgRNA(single guide RNA)组成。与ZFN 和TALEN 相比,CRISPR 系统在基因组编辑方面更加快速、通用且精准[39]。其中,sgRNA 负责识别原间隔区相邻基序(PAM)位点上游的20 个核苷酸序列。当sgRNA 识别出目标DNA 序列后,Cas9 将在PAM和其上游20 bp之间的位点断裂DNA双链,进而激活宿主DNA 非同源末端连接(non‑homologous end joining,NHEJ) 或同源重组(homologous recombination,HR)修复过程(图2)。
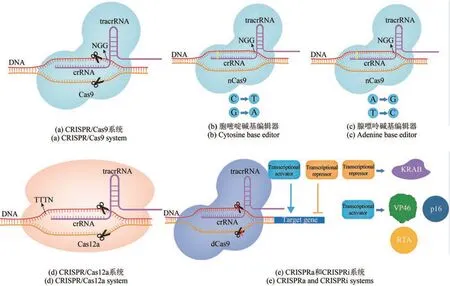
图2 CRISPR/Cas基因组编辑系统作用机制Fig. 2 Mechanism of CRISPR/Cas genome editing systems
2.2 NHEJ和HR修复机制
Cas9 蛋白切割目标靶点产生的DSB,可以通过非同源末端连接(NHEJ)或同源重组(HR)修复。NHEJ 修复可能会随机插入、删除DNA 序列,精准度不高;HR 修复通过同源序列进行,精准度高,是进行精准基因编辑的关键[40]。NHEJ在细胞周期的G1期随机发生,在Ku 蛋白复合物(Ku70/80)的作用下,NHEJ 修复途径可直接使DNA 末端双链断裂进行连接。HR修复发生在DNA复制过程中,同源供体DNA 片段与其特定靶标二者缺一不可[41],能进行精确修复。
基于HR 的原理,Liu 等[38]在丝状真菌里氏木霉中建立了CRISPR/Cas9 系统,发现不同长度同源臂的同源重组效率均在93%以上,实现了有效的外源基因整合。Suzuki 等[42]设计了一种基于CRISPR/Cas9 的同源独立靶向整合(HITI)策略,该方法利用NHEJ途径,在分裂和非分裂细胞中实现靶向敲入,允许通过NHEJ修复将片段整合到基因组中。Clemmensen等[43]将环形供体、线性供体分别转入扩展青霉(Penicillium expansum)中,发现转入环形供体的活转化子数量高于线性供体。这可能是由于环形供体不存在DSB,不能通过NHEJ途径整合到染色体上,从而有利于HR修复[44]。
2.3 Cas9的表达
Cas9 蛋白是CRISPR/Cas9 基因组编辑系统的重要组成部分,具有切割目标靶点的作用[45]。CRISPR/Cas9系统是在细菌或古细菌中发现的,而真菌中不存在Cas9 蛋白[46]。因此,需根据真菌密码子偏好性,重新设计优化Cas9基因序列。此外,需在Cas9基因两端添加SV40等核定位信号[47],以实现Cas9 蛋白的定向表达,完成对真菌基因组的编辑。尽管SV40 广泛应用,但在尖孢镰刀菌(Fusarium oxysporum)[48]和藤仓镰刀菌(Fusarium fujikuroi)[49]等真菌中,需使用其特有的内源核定位信号序列连接Cas9蛋白。
Cas9 蛋白的成功表达依赖于系统所用启动子的类型和强度,选择合适的启动子对于建立高效的CRISPR/Cas9 系统至关重要[50]。在丝状真菌中,主要使用构巢曲霉(Aspergillus nidulans)的trpC[51]、gpdA[52]和tef1[53]组成型启动子表达Cas9蛋白。有研究表明Cas9 的持续表达会产生细胞毒性[54‑55],因此,里氏木霉的cbh1启动子或构巢曲霉的xlnA启动子等诱导型启动子常用于Cas9 的表达。在里氏木霉中,多使用cbh1启动子表达Cas9基因,以减少基因编辑的脱靶效应[38]。经基因组学研究后,工业青霉素生产菌产黄青霉Wisconsin 54‑1255被归属于鲁本斯青霉(Penicillium rubens)[56]。在鲁本斯青霉中,xlnA启动子用于高表达Cas9 蛋白[57]。因此,合适的启动子是优化丝状真菌CRISPR/Cas9系统的方案之一。
2.4 sgRNA的表达
sgRNA 的高效表达是CRISPR/Cas9 系统发挥作用的关键。sgRNA 转录后才能在丝状真菌中发挥作用。U6snRNA 基因序列高度保守[58],因此,RNA 聚合酶Ⅲ型U6启动子是sgRNA 转录的常用启动子之一。在嗜热毁丝霉(Myceliophthora thermophila)中,Liu 等[59]使用U6启动子,以cre-1等基因为靶点,单基因编辑效率高达95%,多基因共编辑效率最高为70%。除U6启动子外,tRNA 启动子也可以用于sgRNA 的转录。通过鲁本斯青霉全基因组测序以及序列注释[60],鉴定出145个tRNA 编码基因。格罗宁根大学的Pohl 等[57]基于序列分析选取了tRNA‑Met 和tRNA‑Leu 两个启动子,成功敲除了与色素合成有关的pks17基因。Song 等[61]报道了在黑曲霉中由内源性tRNA 启动子驱动的gRNA 转录,该启动子包括一个tRNA 基因及其上游的100 bp 序列。进一步研究表明,将表达Cas9 的质粒与线性的gRNA 共转化后,37 个tRNA 启动子中有36 个能够在黑曲霉中转录gRNA[61]。当gRNA 和Cas9 在质粒中表达时,基因突变的效率高达97%[61]。Zheng 等[62]发现5S rRNA 在细胞中高度保守且丰富,他们以黑曲霉的5S rRNA 基因为启动子驱动sgRNA 的转录。黑曲霉的一个多肽合成酶编码基因albA的敲除效率高达96%,远远高于PhU6启动子15%~23%的敲除效率[62]。
除了上述两种启动子,酿酒酵母的SNR52启动子也成功应用于烟曲霉(Aspergillus fumigatus)等丝状真菌的基因组编辑系统的构建[63]。utp25是鲁本斯青霉中U3小核RNA相关蛋白,也可以作为启动子驱动sgRNA的转录[57]。
2.5 CRISPR/Cas系统在丝状真菌中的构建
2.5.1 CRISPR/Cas元件的染色体表达
脱靶效应是丝状真菌基因编辑效率低下的原因之一,CRISPR/Cas 元件的瞬时表达可以降低脱靶现象。诱导型teton启动子已被用于调控烟曲霉菌株中染色体Cas9 的表达[64]。通过将体外转录的sgRNA 传递到表达Cas9 的真菌菌株中,可以实现CRISPR/Cas9的瞬时表达,已被用于构建藤仓镰刀菌[65]等丝状真菌的基因编辑系统(图3)。Xiang等[66]开发了一种自杀载体,该载体将Cas9元件编码在线性修复模板上,该模板包括一个与靶位点同源的标记基因;Cas9 切割靶基因时,标记基因可以在目标靶点重组,Cas9 则会降解,该方法已用于球毛壳菌(Chaetomium globosum)的基因组编辑。

图3 在丝状真菌中构建的CRISPR/Cas系统Fig. 3 The CRISPR/Cas system constructed in filamentous fungi
2.5.2 自主复制质粒
在丝状真菌中,大多数环状质粒没有在染色体外维持和复制的能力,构巢曲霉的AMA1 序列(autonomous replicator derived fromAspergillus nidulans,曲霉自主复制因子)为质粒在丝状真菌染色体外进行自主复制提供了可能[67]。由于高表达的Cas9 和多拷贝的sgRNA,AMA1 质粒可以显著提高转化效率;在非选择性条件下的传代培养可以去除转化子中带有AMA1 的质粒,实现筛选标记的循环使用(图3)[68]。利用AMA1 质粒在鲁本斯青霉[57]、扩展青霉[43]和米曲霉(Aspergillus oryzae)[68]等丝状真菌中实现了高效的基因组编辑。
AMA1 质粒丢失的难易程度取决于真菌种类。Nielsen 等[69]的研究表明一些菌株可能需要多轮的非选择性生长才会失去所有的AMA1 质粒。使用标记基因pyrG[70]或条件致死基因Aoace2[71]等的反向选择策略可以快速去除转化子中的AMA1质粒。
2.5.3 核糖核蛋白复合物递送
核糖核蛋白(ribonucleoprotein,RNP)复合物由Cas 蛋白和体外转录的gRNA 组成,可以递送至丝状真菌细胞内,实现基因组编辑(图3)。RNP 转化无需优化CRISPR/Cas 表达盒,减少了评估启动子转录效率等构建步骤,且RNP 只是短暂存在于体内,降低了脱靶的风险[72]。然而,RNP 介导的编辑效率可能会受到sgRNA 不稳定和Cas9 转运效率低的限制[57]。RNP 已经在里氏木霉[73]、斜卧青霉(Penicillium decumbens)[74]、波兰青霉(Penicillium polonicum)[75]、层出镰刀菌(Fusarium proliferatum)[76]等多种真菌中应用。
3 CRISPR/Cas 基因组编辑在丝状真菌中的应用
CRISPR/Cas 系统已用于多种丝状真菌的基因编辑。通过敲除天然代谢途径中的基因或在宿主细胞中表达异源基因,有助于解析天然产物生物合成途径、增强目标天然产物表达、削弱或消除毒性产物等,促进丝状真菌的开发与应用。
3.1 CRISPR/Cas9介导的基因敲除
基于CRISPR/Cas9 的基因敲除是进行功能缺失研究和探究宿主次级代谢产物生物合成途径的有效策略。基因敲除后,可以改变菌株表型,消除工业真菌中的真菌毒素BGC 或解析特定基因/结构域的功能等。
鲁本斯青霉是生产青霉素的主要工业菌种,经典菌株改良(classical strain improvement,CSI)积累的突变有助于建立以该菌为底盘菌株的天然产物生产平台[77]。CRISPR系统可显著提高青霉菌的遗传改造效率。在鲁本斯青霉菌的基因编辑系统中,AMA1 质粒会在无筛选压力培养时丢失,可以实现无痕编辑。同时,在测试不同长度的供体DNA(donor DNA,dDNA)同源臂后,显示短同源臂(60 bp)即能实现基因功能缺失。通过破坏与色素合成相关的基因,实现了鲁本斯青霉的表型变化,从而表明在鲁本斯青霉中建立了基于CRISPR/Cas9的基因组编辑系统。
由红曲霉(Monascus purpureus)生产的红曲红被用作食品着色剂,但其产生的肾毒性霉菌毒素(橘霉素)会降低红曲红的产量且具有毒副作用。Liang 等[78]认为橘霉素生物合成存在冗余基因或同工酶。因此,为了避免工业真菌合成橘霉素,需要删除整个BGC 而不是破坏单个基因。Liu等[79]设计了一种双质粒CRISPR/Cas 系统,该系统使用环形供体质粒代替线性DNA 供体片段。双质粒CRISPR/Cas9 系统实现了红曲霉工业菌株中15 kb 的橘霉素BGC 的敲除,获得了稳定的同核突变体。传代培养10轮,在每轮发酵过程中均未检测到橘霉素。与原始菌株红曲红色素值(885.30 U/mL)相比,同核突变体红曲红色素值(926.00 U/mL)增加了4.6%。含AMA1 的质粒可以无痕敲除大片段基因簇,因此,可以继续对M. purpureus工业菌株进行迭代基因组编辑。
黑麦麦角菌(Claviceps purpurea)生产的麦角生物碱可用于治疗偏头痛、高血压,也可以在产妇分娩后帮助子宫收缩,减少流血[80]。Yu 等[81]开发了基于体外组装的RNP 的CRISPR/Cas9 系统,对尿苷生物合成(ura5)、菌丝形态学(rac)和麦角生物碱(easA)产生密切相关的3 个靶基因进行敲除,编辑效率达50%~100%。基于体外组装的RNP 可以有效地删除麦角生物碱途径中涉及的相关基因,有助于阐明麦角生物碱的生物合成途径,促进对麦角菌的代谢工程改造,提高麦角生物碱合成效率。
固醇14α‑去甲基化酶(Cyp51)可以催化去除固醇前体中的14α‑甲基基团以转化为麦角固醇[82]。麦角固醇参与多种真菌调控和发育过程,在真菌细胞膜的渗透性和流动性方面发挥重要作用[83]。唑类抗真菌药通过抑制Cyp51酶削弱其前体的去甲基化、阻断麦角固醇的合成,导致细胞内甲基化固醇的积累,从而影响/破坏真菌膜的流动性,抑制细胞增殖[84]。临床上,曲霉属真菌会引起侵袭性曲霉病(invasive aspergillosis,IA)[85]。Pérez‑Cantero等[86]构建了具有不同遗传背景和不同唑类敏感性的cyp51A和cyp51B单敲除突变体。cyp51的单基因缺失导致伏立康唑最低抑菌浓度值低于其流行病学临界值[86]。
赤霉酸(gibberellic acids,GA)是一类四环二萜类化合物,在植物生长调节中起着重要作用,但其生物合成基因并不成簇分布。在藤仓镰刀菌中,GA 的生物合成主要分为牻牛儿基牻牛儿基焦磷酸(geranylgeranyl diphosphate,GGPP)、GA12‑醛和一系列不同GA 类似物3 个部分。GA4 和GA7的混合物与传统使用的GA3 相比,效果更加温和。但在GA 天然代谢途径中,GA4 和GA7 会通过细胞色素P450 单加氧酶基因P450-3进一步转化为GA1 与GA3。Shi 等[49]利用CRISPR/Cas9 系统重构藤仓镰刀菌中GAs 生物合成途径。首先破坏了P450-3基因以阻断GA3 的合成;在P450-3缺失菌株基础上,分别对两个关键基因Cps/Ks和截短的HMGR(tHMGR) 进行过表达。与原始菌株(88.38 mg/L)相比,代谢途径重构的菌株中GA4/GA7 混合物的产量达716.37 mg/L,提高了约7 倍。很多次级代谢产物的基因并不成簇排列,CRISPR/Cas 系统可以对代谢途径中的基因进行敲除或过表达,重构代谢途径和调控网络,从而获得目的产物并提高其产量。
Zhang 等[87]使用CRISPR/Cas9 破坏了黑曲霉中的pyrG和kusA基因后,与对照菌株的产量[(16.98±1.91) g/L]相比,突变菌株的柠檬酸产量[(33.59±3.24) g/L]提高了2.17 倍,表明抑制尿苷/嘧啶的合成可促进柠檬酸的生产。基于RNP 的CRISPR/Cas9系统已经成功应用于黑曲霉,通过该系统在工程菌株中破坏和过量表达多个琥珀酸合成相关基因,并优化温度、pH 等条件,显著提高了琥珀酸的产量[88]。
基于CRISPR/Cas 的基因敲除菌株已用于解析次级代谢产物途径中的单个合成关键酶,并阐明其中间产物的结构与合成途径。Brasiliamides 是由巴西青霉(Penicillium brasilianum)合成的一类含有哌嗪的生物碱,具有多种药理活性[89]。Yuan 等[90]通过删除组蛋白去乙酰化酶激活Brasiliamides 的合成。根据对突变体非核糖体多肽合成酶(nonribosomal peptide synthetase,NRPS)的研究,他们提出了一条新的Brasiliamides生物合成途径,为后续Brasiliamides的大量生产提供了新思路。
3.2 CRISPR/Cas9介导的基因敲入
卡泊芬净是首个获得FDA批准的棘白菌素类抗真菌剂,对念珠菌属和曲霉菌属真菌有良好的临床活性[91]。Glarea lozoyensis可以合成卡泊芬净的重要前体纽莫康定B0(Pneumocandin B0)。在发酵过程中,由于脯氨酸羟化酶基因gloF的区域选择性不高,G. lozoyensis会产生PC0等纽莫康定B0的类似物。棘白菌素B生物合成中的脯氨酸羟化酶Ap‑Hty具有高区域选择性[92]。Wei等[93]采用CRISPR/Cas9策略表达ap-htyE并成功取代gloF,突变菌株保留了合成纽莫康定B0 的能力,但消除了合成纽莫康定C0 的能力。通过替换关键酶,可以减少纽莫康定C0的分离步骤,具有工业应用的潜力。
来自青霉属和曲霉属的部分真菌已经成为真菌BGC 的异源表达宿主[94]。McLean 等[77]使用鲁本斯青霉表达来自橘青霉的美伐他汀BGC(mlcA、mlcB、mlcC、mlcD、mlcE、mlcF、mlcG、mlcH、mlcR),同时表达一个细胞色素P450基因,将美伐他汀转化为普伐他汀。在中试规模下,普伐他汀发酵产量超过6 g/L。他们使用的鲁本斯青霉删除了青霉素合成基因簇,该宿主经过多轮菌株改良,释放了发酵条件下合成次级代谢产物的潜力。这项工作降低了异源宿主合成天然产物的优化成本,为表达其他天然产物合成基因簇提供了基础。
3(2H)‑呋喃酮是一些活性天然产物的重要组成部分[95]。Setosusin 是一种真菌类二萜,具有独特的螺‑稠合3(2H)‑呋喃酮结构。Wei 等[96]在真菌杜氏曲霉(Aspergillus duricaulis)中鉴定了与Setosusin 相关的BGC,通过在米曲霉中异源重组相关酶基因,阐明了其生物合成途径。Katayama等[68,97]在半AMA1 载体上提供了可诱导的反选择标记Aoace2,实现了米曲霉的无标记整合。
内消旋半乳糖二酸是一种六元酸,可以作为护肤品成分使用,也可以用于生产聚合物,它可通过果胶的主要成分D‑半乳糖醛酸氧化生成。黑曲霉生产的果胶酶效率高,适合于转化富含果胶的生物质。Kuivanen 等[98]使用CRISPR/Cas9 与体外合成sgRNA 删除了内消旋半乳糖二酸相关的基因,中断内消旋半乳糖二酸代谢,构建了D‑半乳糖醛酸分解代谢与异源糖醛酸脱氢酶表达相结合的工程化黑曲霉菌株,最终可以通过D‑半乳糖醛酸合成内消旋半乳糖二酸。Dong 等[99]利用CRISPR/Cas9 介导的多拷贝敲入表达策略,在黑曲霉中高效表达了来自嗜热毁丝霉的高活性海藻糖酶(MthT),最终,工程黑曲霉催化海藻糖水解,酶滴度可达1698.83 U/mL。
除青霉属和曲霉属之外,多种其他真菌也被开发为异源天然产物生产平台。麦角酸(LA)和二氢麦角酸(DHLA)是许多药物的先导化合物,可用于治疗痴呆、偏头痛、高催乳素血症和其他疾病[100‑101]。但它们通常是麦角生物碱和二氢麦角生物碱合成途径中的中间代谢产物,难以获取。褐色绿僵菌(Metarhizium brunneum)可以天然合成多种LA 酰胺,最终合成麦角酸α‑羟乙胺(LAH)以及少量的麦角新碱[102]。Davis 等[103]通过RNP 策略在褐色绿僵菌中沉默麦角生物碱途径,并异源表达外源基因,合成了麦角酸和二氢麦角酸,且麦角酸(86.9%)和二氢麦角酸(72.8%)的相对产量远高于已有的工程烟曲霉菌株(分别为2.6%和2.0%)。
表1 总结了丝状真菌中CRISPR/Cas 基因组编辑系统的应用情况。
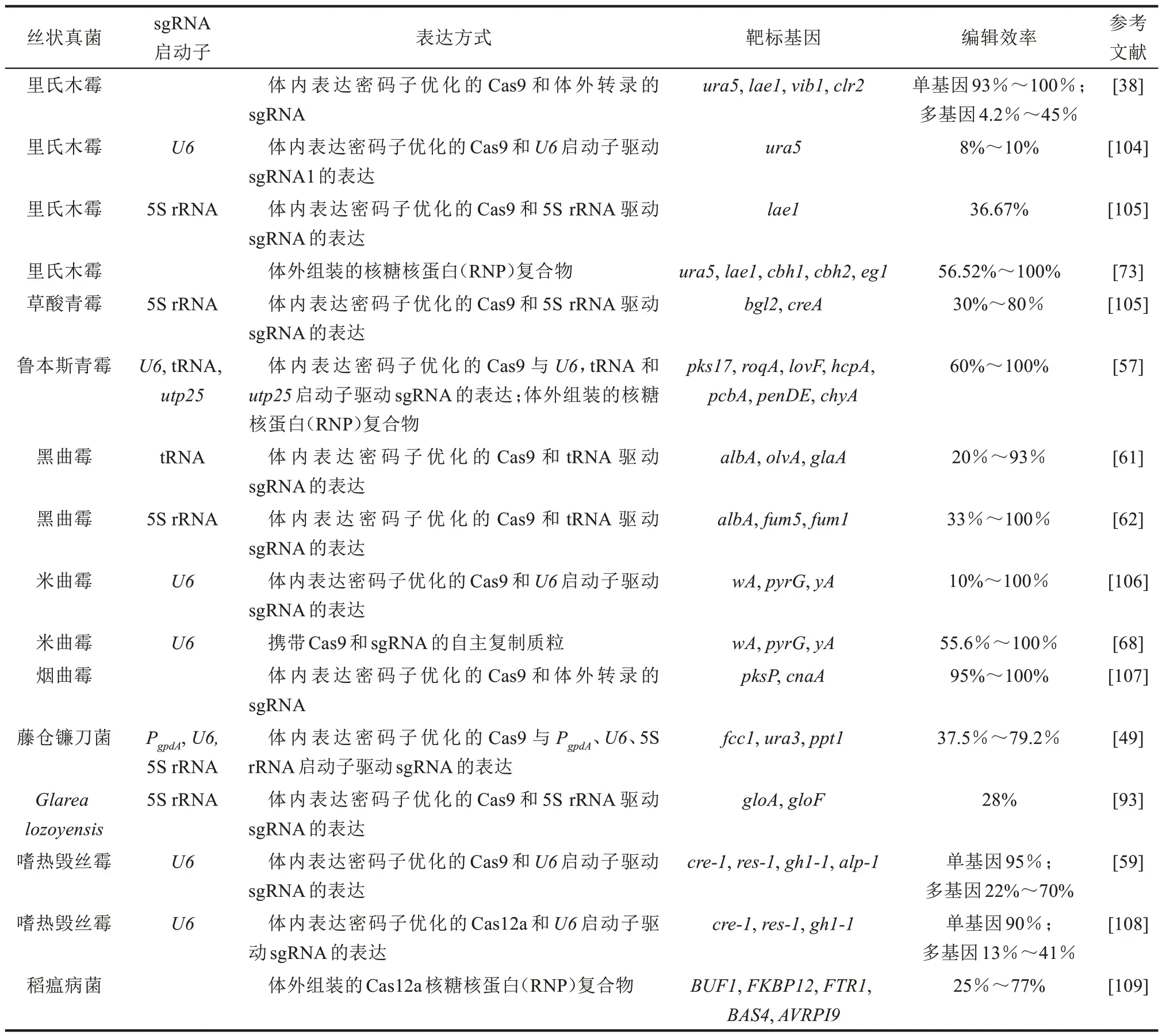
表1 丝状真菌中CRISPR/Cas基因组编辑系统的应用Table 1 Application of CRISPR/Cas genome editing system in filamentous fungi
3.3 CRISPRa和CRISPRi系统
CRISPRa 通过核酸内切酶失活的Cas9(nuclease‑dead mutants of Cas9,dCas9)连接转录激活子,实现基因组特定位点的转录[110]。CRISPRi 通过dCas9 与转录抑制子连接,在gRNA引导下,结合到靶基因位点,抑制转录起始,沉默靶基因的表达[111]。Roux 等[112]在构巢曲霉中构建了首个CRISPRa 系统,通过激活micA基因,提高了微呋喃酮的产量,并通过多基因CRISPRa 鉴定出新代谢产物脱氢微呋喃酮。丝状真菌有能力合成多种高价值次级代谢产物,但大多数BGC 是沉默的。CRISPRa 与次级代谢产物BGC 的表达激活有助于获取活性天然产物。Li 等[113]使用CRISPRi 对黑曲霉中的表观遗传效应进行了研究。通过dCas9 作用于内源性组蛋白乙酰化酶GcnE 和组蛋白去乙酰化酶HosA 和RpdA,抑制了BGC 的转录,但未发现表型的变化和新代谢产物的产生。
3.4 基于CRISPR/Cas的高通量筛选
与ZFN和TALEN相比,CRISPR/Cas系统可以用于高通量正向遗传筛选[114]。sgRNA 是CRISPR/Cas系统的关键元件,传统的构建sgRNA文库方法是基于已知的全基因组序列设计并合成上万个sgRNA,但昂贵的成本以及未知的真菌全基因组阻碍了基于CRISPR/Cas9的全基因组筛选应用。Li等[115]开发了酶切/结合农杆菌介导的转化方法(restriction/ligation coupled with Agrobacterium‑mediated transformation,RELATe),在人类真菌病原体新型隐球菌(Cryptococcus neoformans)中创建的sgRNA 文库可以靶向基因组中超过98%的蛋白质编码基因。新型隐球菌可以穿透血脑屏障,在免疫功能低下的人群中会引发中枢神经系统(central nervous system,CNS)感染。通过高通量功能筛选确定了142 个与发病机制相关的潜在基因。研究人员选择SFP1和WDR1两个基因进行了敲除,结果表明SFP1和WDR1是新型隐球菌渗透血脑屏障的必需基因。新开发的RELATe方法降低了高通量筛选的成本,与CRISPR/Cas 系统结合可以用于阐明真菌功能基因。
有效的高通量筛选策略可以从突变体库中分离所需的突变体,进而使用CRISPR/Cas 系统对其进行遗传改造。Luu 等[116]在里氏木霉中开发了一种基于液滴微流控技术的绿色荧光蛋白表达的新方法。他们将转化绿色荧光蛋白的里氏木霉原生质体封装并在液滴中孵育24 小时,通过高通量筛选液滴,最终收集到一个转化文库,标记基因转化率达96%以上。与传统转化相比,该方法使原生质体再生时间大大缩短,再生频率提高了8 倍,筛选速度可达每分钟8000 液滴。该方法提高了筛选效率,有望应用于其他丝状真菌转化子的筛选,加快遗传转化速度。
3.5 丝状真菌CRISPR/Cas9 系统面临的问题及可能的解决方案
3.5.1 脱靶效应
在基因编辑过程中,sgRNA 引导Cas9 至与靶位点相似核苷酸序列结合,会造成潜在的脱靶效应。为了减少脱靶的出现,需要设计序列特异性较高的sgRNA。近年来,已经有多个用于预测脱靶位点并为sgRNA 进行评估的网站,如E‑CRISP[117]、CHOPCHOP[118]、Cas‑OFFinder[119]等。此外,研究人员开发了多种方法提高Cas9 切割的特异性,如构建Cas9‑sgRNA 核糖核蛋白复合物进入细胞[120]、使用Cas12a 替换Cas9[121]等。设计适宜的sgRNA 是限制脱靶效应的关键因素。sgRNA 邻近原型间隔区相邻基序(PAM)的10~12 bp 决定了Cas9 切割特异性[122]。如果基因组含有与靶点sgRNA 相似性过高的序列,会导致Cas9 在非靶向基因位点的非预期DNA切割[123]。因此,设计独特适宜的sgRNA,能够降低Cas9脱靶切割的可能性。新发现的Cas12b[124]、Cas13[125]、Cas14[126]等核酸酶的应用有望在解决脱靶效应等问题中发挥作用。
3.5.2 转化效率低
丝状真菌较厚的细胞壁是外源基因片段进入细胞的主要障碍。破除细胞壁后的原生质体可以有效吸收外源DNA,但是原生质体的低再生率降低了丝状真菌的转化效率。Han 等[127]引入并优化了一种微流体细胞膜变形方法,这个方法使用快速的细胞机械变形来产生瞬时膜孔,不但能将sgRNA 和Cas9 传递至难以转染的淋巴瘤细胞和胚胎干细胞中,还能保持较高的细胞活力。丝状真菌是多核微生物,在菌丝和孢子中含有数量不定的细胞核[128]。即使其中一个细胞核的基因被破坏,其余细胞核的基因仍能保持完好,这也是转化菌株假阳性高的原因之一。中国科学院分子植物科学卓越创新中心周志华课题组[73]在多核孢子真菌米曲霉中开发了基于RNP的CRISPR系统,通过加入化学试剂肌醇或苯菌灵,增加了单核原生质体的形成,无需进行后期的孢子分离步骤,提高了单核菌株的形成率,最高转化效率达86.67%。通过加入Triton X‑100、吐温‑80等表面活性剂,可以促进RNP复合物进入细胞,提高转化效率。周志华团队[73]在里氏木霉原生质体转化中加入Triton X‑100,菌落形成单位(colony forming units,CFU)较之前增加3.33倍。
3.6 丝状真菌中的CRISPR/Cas12a基因组编辑
使用Cas9 进行基因编辑会受到其5'‑NGG‑3'PAM 序列的限制[129],位点特异性不高。因此,引入依赖于其他PAM 序列的RNA 引导的核酸酶能够更精确靶向基因组位点。毛螺菌科细菌的Cas12a(也被称为Cpf1)采用由5'‑TTTN‑3'组成的PAM 序列(图2)[129]。Abdulrachman 等[121]在棘孢曲霉(Aspergillus aculeatus)TBRC 277 中开发了基于AMA1的CRISPR/Cas12a表达载体,并建立了三种不同的靶向pyrG基因的引导crRNA,证明了Cas12a 能够诱导位点特异性双链断裂。Huang等[109]在真菌病原体稻瘟病菌(Magnaporthe oryzae)中建立了基于Cas12a核糖核蛋白的基因组编辑系统。BUF1编码真菌黑色素生物合成所需的三羟基萘还原酶,研究人员设计了两种靶向BUF1位点的gRNA,并与Cas12a 蛋白复合为RNP 转化原生质体,BUF1位点的编辑率达到77%。Cas12a提高了基因编辑精度,将在丝状真菌沉默基因簇挖掘和次级代谢产物研究中发挥作用。
破坏NHEJ 修复是提高基因组精准编辑效率的有效策略。美国堪萨斯州立大学Huang 等[109]在稻瘟病菌中发现CRISPR/Cas诱导的DNA双链断裂存在多种修复途径。在丝状真菌中,DNA 双链断裂修复主要由NHEJ 和HR 两种途径,但仍存在微同源介导的末端连接(microhomology‑mediated end‑joining,MMEJ)[130]和单链退火(single strand annealing,SSA)[131]两条修复途径。研究人员通过PCR、Sanger测序和纳米孔长读值测序技术的组合,发现在进行CRISPR/Cas12a 介导的基因编辑后,Ku80 缺失菌株中发生了较野生菌株中更大片段的DNA缺失,这是MMEJ介导的DNA突变的特征。MMEJ 等修复途径可能会产生不可预测的基因组变异,从而引发CRISPR/Cas 系统的安全性问题。
3.7 基于CRISPR的丝状真菌碱基编辑系统
碱基编辑(base editing,BE)分为胞嘧啶碱基编辑器(cytosine base editor,CBE)和腺嘌呤碱基编辑器(adenine base editor,ABE)(图2)。CRISPR 介导的碱基编辑可以在靶基因组位点上转换碱基,具有无需双链断裂、无需供体DNA 以及同源定向修复等优点。第三代碱基编辑器(third‑generation base editor,BE3)是最常用的碱基编辑器系统,由dCas9 或Cas9‑D10A 切口酶(D10A nickase,nCas9)与大鼠胞苷脱氨酶(rat cytidine deaminase,rAPOBEC1)和尿嘧啶糖基化酶抑制剂(uracil glycosylase inhibitor,UGI)组成,可以实现胞嘧啶(C)转化为胸腺嘧啶(T)或腺嘌呤(A)转化为鸟嘌呤(G)[132]。华南理工大学Huang等[133]在黑曲霉中应用了胞嘧啶碱基编辑器,通过单碱基编辑诱导无义突变,灭活了尿苷相关基因pyrG和色素相关基因fwnA,效率为47.36%~100%;对于非表型基因prtT的单碱基编辑效率达60%。该系统为研究黑曲霉以及其他丝状真菌提供了便捷的工具。天津工业生物技术研究所田朝光团队[134]在嗜热毁丝霉中构建了进化型载脂蛋白B mRNA 编辑酶催化亚基1(APOBEC1)胞嘧啶碱基编辑器4max(Mtevo‑BE4max)、噬菌体Mu Gam 蛋白胞嘧啶碱基编辑器4max(MtGAM‑BE4max)、进化型CDA1脱氨酶胞嘧啶碱基编辑器(Mtevo‑CDA1)等三个新的胞嘧啶碱基编辑器,并成功编辑了amdS、cre-1和Mtclr-2三个靶基因。研究人员深入研究了Mtclr-2的功能,发现其与分生孢子的产生、菌丝生长和菌落形态的维持存在密切关系。Mtevo‑CDA1 的碱基编辑效率可达92.6%。由于碱基编辑不需要双链断裂和供体片段,可以弥补CRISPR/Cas9 的不足。但在水稻中应用BE3 后,显示其诱导了大量的全基因组脱靶突变[135]。未来,仍需对碱基编辑系统组成元件和可靠性进行优化和验证[136‑137]。
4 总结与展望
自CRISPR/Cas 基因组编辑系统应用于丝状真菌以来,研究人员对Cas9 核酸酶、sgRNA 的表达以及CRISPR/Cas 系统的递送方式不断优化,在模式或非模式丝状真菌中建立了多个CRISPR/Cas 系统。CRISPR/Cas9系统是目前应用最广泛的基因组编辑系统,但也存在Cas9核酸酶毒性、脱靶效应、转化率低等问题。Cas12a、Cas12b、Cas13、Cas14等核酸酶的应用有望解决这些问题。
本文聚焦于丝状真菌天然产物在医药领域的开发与应用。纽莫康定B0 和他汀类药物等活性天然产物在野生菌株中存在生物合成途径基因表达强度不高、副产物代谢途径影响等问题。通过CRISPR/Cas 进行基因缺失或异源表达,可以探索代谢产物的BGC、改造次级代谢产物生物合成基因、促进丝状真菌细胞工厂的构建。CRISPR/Cas技术可以进行多基因共编辑,调控副产物合成途径中的多种酶基因,增强目标产物的积累并减少毒性物质的产生。CRISPR/Cas 技术还可以通过选择功能元件、重构底盘细胞、优化代谢途径等一系列策略实现丝状真菌次级代谢产物的高产,从而加速丝状真菌功能基因组学和次级代谢产物的研究和工业应用。
