光酶催化合成进展
2023-09-16明阳陈彬黄小强
明阳,陈彬,黄小强
(南京大学化学化工学院,南京大学化学与生物医药创新研究院,配位化学国家重点实验室,江苏 南京 210023)

自然界中的酶是细胞产生的天然催化剂,由于酶活性口袋独特的空间结构,它可以降低反应活化能并调控反应过程,从而可以在温和条件下催化高度化学选择性、区域选择性和立体选择性的反应[1]。得益于微生物学、分子生物学和合成生物学等相关领域的高速发展,许多具有优异催化性能的酶已经可以市场化,大大扩展了生物催化工具箱[2]。以酶催化为基石的生物制造可以显著降低成本和能源消耗,还具有优异的反应活性和选择性,因此酶广泛用于食品和动物饲料、日化、制药和生物技术研发等行业[3‑5]。最近的一份统计报告显示,2021 年全球酶市场价值约58 亿美元,预计到2031 年将达102 亿美元[6]。而在学术研究领域,化学家们对反应选择性,尤其是立体选择性的追求越来越高。酶催化由于具有绿色可持续、活性高与选择性可进化、可调控的优点,所以其被视为一个解决传统合成所面临的环境、效率和立体控制等问题的潜在完美方案[7‑8]。然而,相对于传统化学催化剂,现有酶催化的反应类型相对有限,难以满足绿色生物合成的需求。因此,如何通过扩展酶的催化功能来实现更多更广泛的生物合成,是当下科学前沿的热点问题[9]。
太阳能是一种环保无污染、丰富且可再生的清洁能源,因此被视为一种绿色化学合成的“理想试剂”[10]。1912 年,有机光化学的先驱人物Giacomo Ciamician 最早阐述了这一观点[11],在题为《未来的光化学》的文章中提到:一个新的、对环境更负责任的化学工业,可以用清洁、经济、高效的光化学转化取代高能合成工艺,并由此带来巨大的生态效益。就像植物能利用阳光作为可再生能源将CO2和水转化为有机化合物,光化学合成相较于传统合成的优势在于更清洁、可持续。此外,新型高效光催化剂的开发[12‑13],丰富了光催化的机理途径[14],进而推动了光催化水裂解制氢[15]、CO2还原生产燃料[16]、降解有机污染物[17‑18]以及选择性有机合成[19‑21]等领域的发展。在过去的十余年中,可见光催化与合成化学的交叉融合已经成为了一个非常重要的研究领域。然而,光诱导所产生的高能有机中间体通常面临着活性过高、反应性难以调控、副反应较多等问题[22‑24]。因此,如何恰当地设计光催化合成体系,从而合理地避免副反应发生,提高反应的选择性,是目前光催化有机合成的难点。
光酶催化能够一定程度上解决可见光催化和酶催化分别面临的问题,同时通过整合各自的优势,实现高附加值化合物的高效、可持续绿色制造[25‑28]。一方面,基于可见光催化在温和条件下产生的活泼化学中间体,光酶催化不仅能够利用光再生辅因子来发挥酶的天然活性,还能引发酶的非天然反应活性,获得新酶催化功能;另一方面,利用酶催化的高选择性和可定向进化的特性[29‑30],光酶催化能够调控光引发的活泼反应中间体,从而为光化学领域的立体化学控制难题提供新的解决方案。自然界中被人们所揭示的天然光酶催化合成系统有四类,包括:光合系统[31‑32]、光裂合酶[33]、原叶绿素酸酯氧化还原酶[34]和脂肪酸光脱羧酶[35‑36]。近年来,得益于多学科技术的交叉融合,非天然的光酶催化发展迅速,在合成领域展现出了重要的应用潜力。光酶催化目前已成功用于催化碳碳、碳氮、碳氧、碳卤成键等重要的生化反应,在催化活性、反应选择性、底物谱拓展等方面表现出其独有的优势[8]。
本文综述光酶催化合成领域的最新研究进展,根据光与酶的结合模式,将本文分成四部分内容讨论(图1):光氧化还原实现辅因子再生;光催化剂‑酶的协同或串联;光激发已知酶实现新转化;人工光酶。本文将归纳近年来典型的有关光酶催化合成的研究报道,重点分析光酶催化反应的化学机制和实现新生物转化的策略,通过分析该领域当下面临的瓶颈,进一步展望光酶催化未来的发展方向。
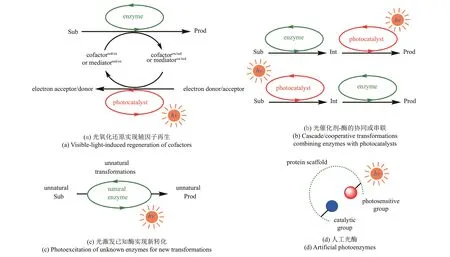
图1 光酶催化模式图Sub-底物;Prod-产物;Int-中间体Fig. 1 Photobiocatalytic modes diagramSub-substrate; Prod-product; Int-intermediate
1 光氧化还原实现辅因子再生
通过光氧化还原催化实现酶促反应所需辅因子的再生,是光酶结合最直观的系统,也是最容易实现、早期报道最多的系统[37],其意义在于替代成本高昂的天然辅因子,或简化电子传递链从而实现更高效的反应。如图1(a)所示,该耦合系统有5 个组成部分:底物/产物、氧化还原酶(redox enzyme)、辅因子(cofactor)/氧化还原介质(mediator)、光催化剂(photocatalyst)以及电子牺牲供体/受体。通常,该系统可以看作由协同的酶氧化还原循环和光氧化还原循环组成。以光催化循环实现辅因子的还原再生为例:首先,光催化剂被光激发,从牺牲电子供体中捕获电子或氢原子;随后,电子从还原态光催化剂直接或间接转移至辅因子,实现辅因子的还原再生;最后,氧化还原酶利用再生的辅因子,将底物还原为对应产物[38]。目前常见的牺牲电子供体包括叔胺[如三乙醇胺(TEOA)、乙二胺四乙酸(EDTA)]、有机酸(如甲酸、抗坏血酸)、含硫化合物(如甲硫氨酸、联二亚硫酸盐)、两性离子缓冲液[2‑吗啉乙磺酸(MES)、4‑羟乙基哌嗪乙磺酸(HEPES)]等;而光催化剂的种类更加多样,包括各种金属有机络合物、有机染料(如卟啉‑叶绿素、黄嘌呤、黄素)和纳米材料[如量子点(QD)、氮化碳(CN)、碳点(CD)]等;目前报道过的辅因子/介质主要包括烟酰胺腺嘌呤二核苷酸/烟酰胺腺嘌呤二核苷酸磷酸NAD(P)H、黄素(flavins,如FMN/FAD)、甲基紫精(methyl viologen, MV2+)、铑络合物等[39](图2)。目前已有许多综述总结了有关研究[37‑41],因此本文仅介绍一些近期报道的典型例子。
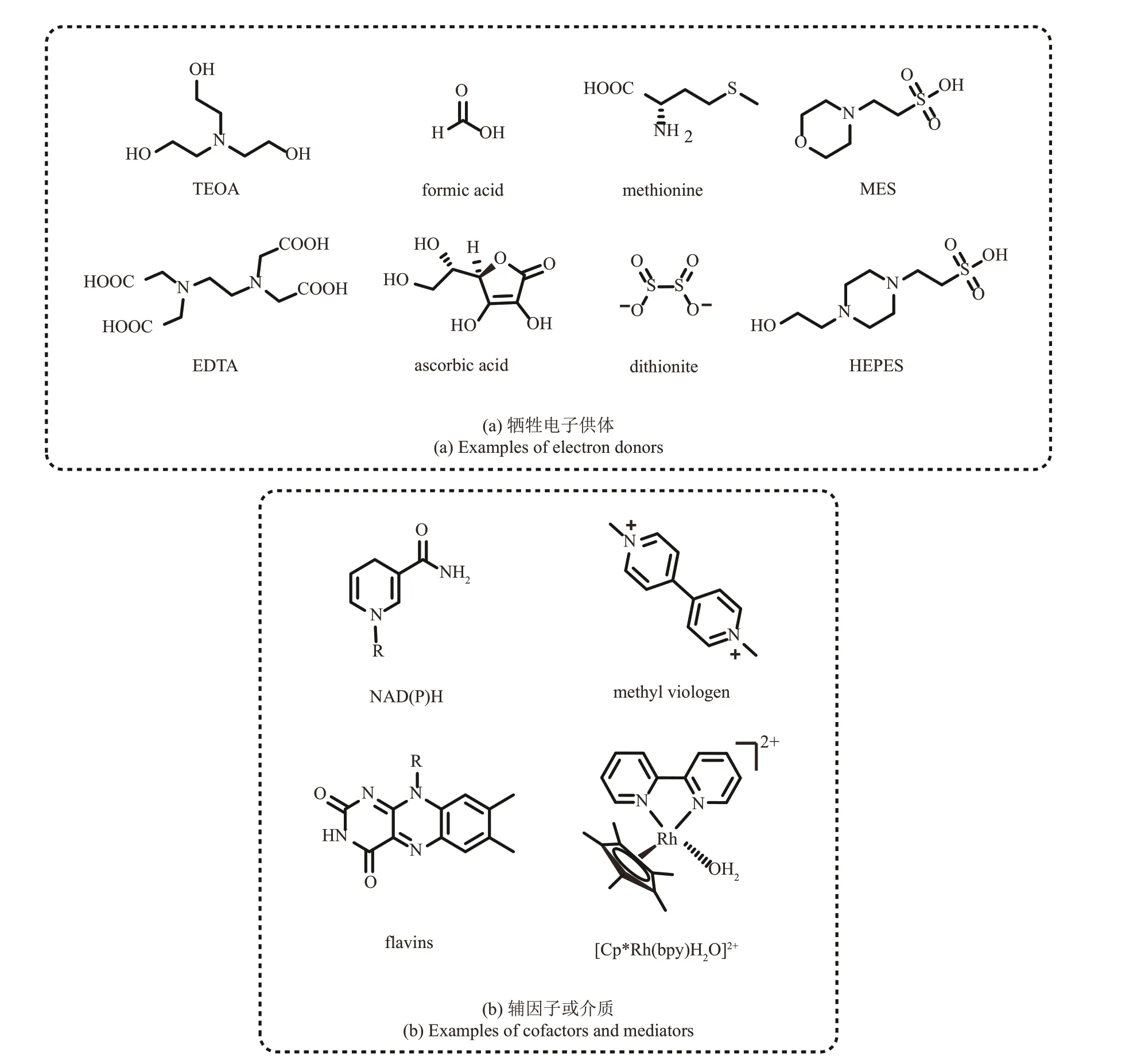
图2 光氧化还原实现辅因子再生系统中常见的牺牲电子供体、辅因子或介质Fig. 2 Common sacrificial electron donors and cofactors/mediators in photoredox‑enabled cofactor regeneration system
2018 年,Paul 和Park 等[42]报告了使用光驱动氮掺杂碳纳米点(N‑CDs)再生辅因子NADH 及其类似物,并用于老黄酶(old yellow enzyme,OYE)催化的不对称碳碳双键的还原反应。由N‑CDs产生的光激发电子还原铑络合物(Mox),还原态的铑络合物Mred将基质中的辅因子NAD+还原进而将氢负提供给老黄酶中的辅基(黄素单核苷酸FMN),完成酶活化底物的立体选择性还原。同年,Kottke 等[43]使用蓝光和牺牲还原供体EDTA再生黄素腺嘌呤二核苷酸(FADH2),开发了一例光化学驱动的黄素依赖卤代酶催化的色氨酸5‑位氯化反应。该系统通过照射酶结合的黄素而不是游离黄素,避免了游离黄素之间的无效电子循环,提高了电子传递效率。
在光合系统中,锚定在类囊体膜上的铁氧还蛋白将电子传递给NADP+还原酶(FNR),实现NADPH 的高效再生。受此启发,整合天然酶与半导体光催化剂而构建的光‑酶耦合催化系统(PECCS),在清洁能源转化和大宗化学品合成方面具有巨大的前景。姜忠义/石家福团队[44]将铑络合物([Cp*Rh(bpy)H2O]2+)通过单宁酸/聚乙烯亚胺(TA/PEI)黏合层锚定在聚合物氮化碳(PCN)上,获得了PCN@TA/PEI‑Rh 这种核壳结构的光催化剂。在可见光照射下,电子从PCN 核被激发,然后通过TA/PEI 壳转移,最后被表面锚定的铑络合物捕获,用于NADH 的再生。核壳结构促进了电子转移,将电子利用效率提高了约1.3 倍。将该NADH 再生系统与甲酸脱氢酶耦合,实现了从二氧化碳到甲酸的生产。之后,他们进一步优化了该核壳的光催化体系,以石墨碳氮化物(GCN)内核作为光敏剂,将铑氢络合物包埋在二氧化钛涂层(MH/TiO2)中作为氧化还原介质,用于乙醇脱氢酶(alcohol dehydrogenase,ADH)催化的乙醇生产[45]。其中MH/TiO2涂层起到两个重要作用:①保护乙醇脱氢酶不被涂层中的GCN 核和MH 影响而失活;②允许电子从GCN转移到NAD+,然后在ADH催化下转移到甲醛。该光酶系统能以(1.78±0.21)μmol/(min·mgADH)的速率产生甲醇,比不含涂层的ADH‑GCN 系统高了420%。
除了核壳结构,受类囊体颗粒堆叠结构的启发,姜忠义/石家福团队[46]还设计了堆叠管状聚合氮化碳(st‑PCN*)负载氯过氧化物酶(chloroperoxidase, CPO)的光酶耦合系统--st‑PCN*@CPO‑PECCS,前者既是光氧化还原再生H2O2的模块,也是酶的载体。该结构克服了传统光酶耦合系统中由于使用游离的酶所导致的辅因子扩散距离长、利用效率低的缺点,使整体光酶耦合催化效率显著提高。在模拟阳光照射下,该系统可以向CPO 原位、连续和可控地供应H2O2,以完成后续的单氯二酮(MCD)转化为二氯二酮(DCD)的氧化氯化反应,MCD 的最终转化率可达39.5%。该系统的初始反应速率(MCD 的转化速率)可达0.29 mmol/(L·h),是具有非堆叠结构对应PECCS的20倍以上。进一步,姜忠义/石家福团队[47]开发了一种基于类囊体膜的胶囊结构的光‑酶耦合催化系统(图3),重点增强辅因子穿梭模块的效率,从而提高整体光酶耦合催化的效率。在该系统中,酶和光催化剂分别固定在膜的外表面和内表面上,而NAD+/NADH辅因子被限制在膜中,并在酶和光催化剂之间快速穿梭,NAD(H)的表观和固有穿梭数是非集成PECCS 的8~12 倍。将该系统用于丙醛还原为丙醇的反应,转化频率(TOF)可达(38 000±365) h-1。
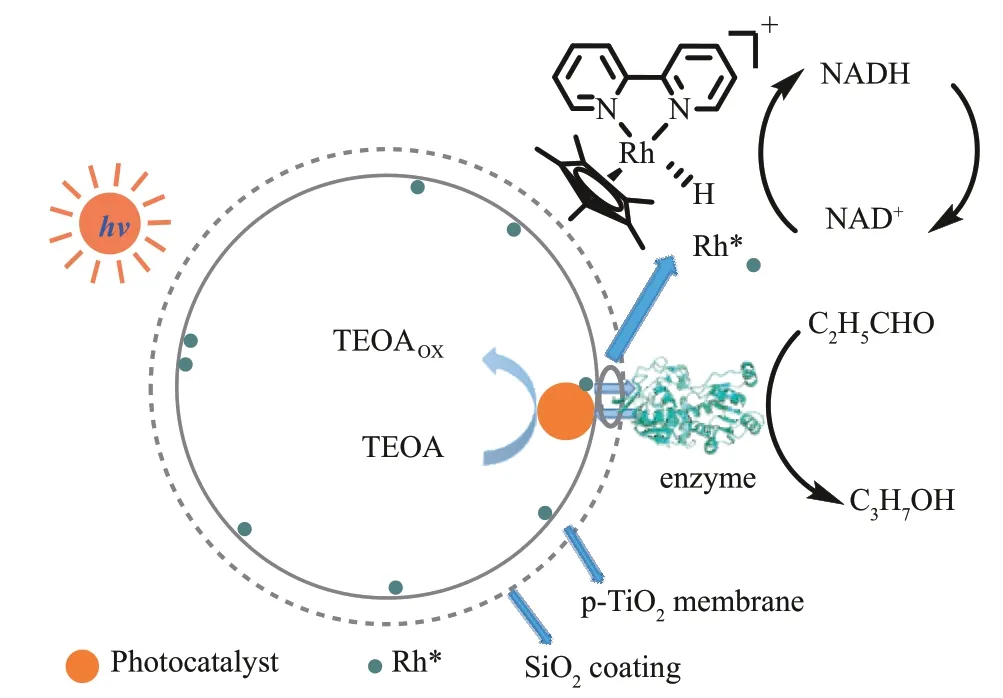
图3 类囊体胶囊启发的光酶耦合系统及辅因子穿梭过程示意图Fig. 3 Illustration of the thylakoid membrane‑inspired capsule (TMC) and the NAD+/NADH shuttling process
2 光催化剂-酶的协同或串联
由于光催化反应通常在室温下进行,并且可以通过光引发的电子转移或能量转移[48]生成水相中稳定的中间体,这使得光与酶的协同或串联组合成为可能。通过光催化剂‑酶的协同或串联,可以设计多种新颖的反应,同时避免分离中间产物的步骤,有利于简化操作并提高效率[49]。但是由于光化学反应和酶催化反应的条件通常不完全相符,且两个体系有可能互相干扰,因此开发动力学匹配、兼容耐受的光酶协同体系[50],或是通过酶固定化等方法避免体系间的不利影响,是开发新型光催化剂‑酶的协同或串联系统的重点。根据外加光催化剂发挥的不同作用,本节分以下四部分内容展开讨论。
2.1 光能量转移实现底物异构化
2018 年,赵惠民、Hartwig 等[51]报道了基于光能量转移和烯烃还原酶的协同催化反应[图4(a)]。作者利用[Ir(dmppy)2(dtbbpy)]PF6或FMN 作为光催化剂,通过光诱导能量转移促进烯烃的E/Z异构化;同时结合烯烃还原酶和烟酰胺辅因子再生系统,实现酶催化优选的E‑构型烯烃的高对映选择性还原,从而富集得到单一构型的还原产物。在此基础上,2020 年赵惠民团队[52]通过利用光氧化还原再生FMNH2的系统,在不使用NADPH 再生系统的情况下(以EDTA 或TEOA 为终端还原剂),也实现了活化烯烃的协同立体汇聚式还原。
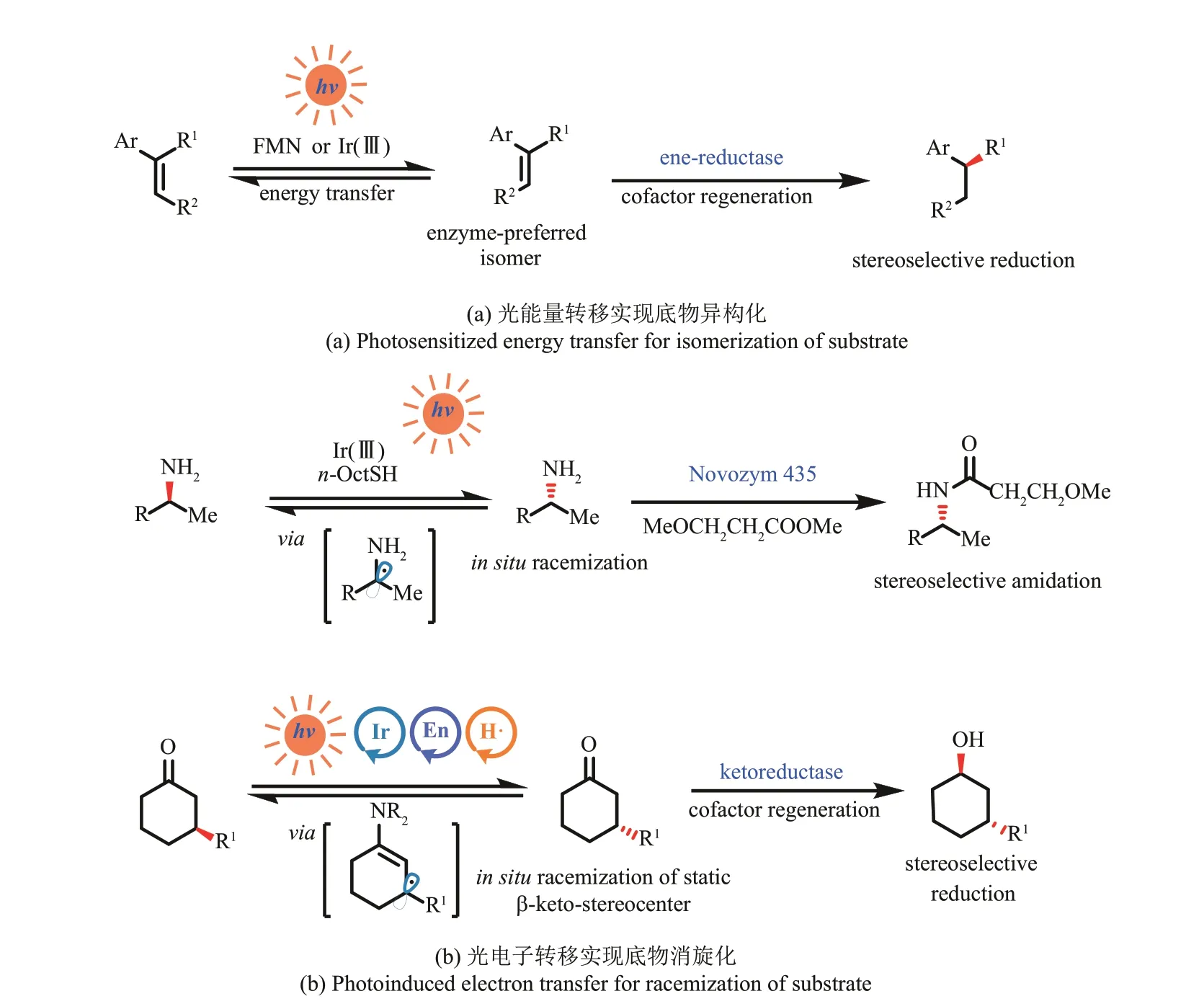
图4 光引发的能量/电子转移生成酶优选底物En-烯胺催化;H·-氢原子转移催化Fig. 4 Convert substrate into enzyme‑preferred ones by photoinduced energy/electron transferEn-enamine catalysis; H·-hydrogen atom transfer catalysis
2.2 光电子转移实现底物消旋化
动态动力学拆分(dynamic kinetic resolution,DKR)依赖于底物对映体间的快速相互转化,结合立体选择性键形成,可以将外消旋底物转化为对映体纯的产物[53‑54]。
2018 年周少林等[55]利用可见光引发的单电子转移(single electron transfer, SET)实现底物消旋,并协同脂肪酶催化的选择性酰胺化,实现了一例温和条件下的胺类化合物的动态动力学拆分[图4(b)上]。机理上,光激发的双环金属化铱络合物首先氧化硫醇,生成相应的硫自由基,通过可逆的氢原子转移(hydrogen atom transfer,HAT)实现氨基邻位的立体中心的快速消旋化。另外,作者使用商品化的脂肪酶Novozyme 435,能够选择性识别某一个构型的胺,在酰基供体存在的条件下,完成动态动力学酰胺化转化。
2020 年MacMillan、Hyster 等[56]结合光‑小分子胺催化的酮羰基β‑C-H 键的消旋和酶催化的对映选择性羰基转化,成功实现了远程惰性C-H 键的动态动力学拆分[图4(b)下]。基于MacMillan前期有关小分子胺催化和光催化的研究[57],作者利用激发态光催化剂单电子氧化烯胺中间体,得到相应的自由基阳离子中间体,从而使得原羰基的惰性β‑C-H 键酸性增强;经由去质子化过程,转变成5π 电子的自由基中间体;该5π 电子中间体为平面型中间体,在后续的氢原子转移重构β‑CH键过程中,完成羰基化合物β‑位静态立体中心的外消旋化。与此同时,协同利用酮还原酶(或转氨酶)选择性识别R-构型的羰基底物,不断地将原料立体选择性地还原为具有双手性中心的醇(或胺)。作者提出的这一光‑化学小分子催化剂结合的策略,构建了一类特有的外消旋化平台,突破了传统上对于静态立体中心的定义,通过与其他催化羰基官能团转化的生物催化过程联用,有望开发更多立体汇聚式转化。
2.3 光催化剂选择性还原酶活化的底物
除了通过光能量/电子转移实现底物的异构化/外消旋化,还可以利用酶活口袋对于底物的氢键活化作用,降低底物还原电势,从而使光催化剂可以选择性还原酶结合的底物,再结合酶活性口袋的立体化学控制能力,开发高选择性的非天然生物催化反应。
2018 年,Hyster 等[58]利用光氧化还原催化剂与酶的结合实现了氧化还原酶新的催化功能[图5(a)]。在可见光照射下,激发态的光催化剂Rose Bengal 将酶口袋中通过氢键结合的底物单电子还原,脱去乙酸根阴离子后生成α‑羰基碳自由基中间体,接着与酶口袋中的NADPH 发生HAT,最终实现烟酰胺依赖的双键还原酶催化的对映选择性脱乙酰氧基反应。此外,以曙红Y 为光催化剂,酮还原酶还可以催化α‑溴酰胺的自由基脱卤化反应。该策略展示了通过光氧化还原催化与生物催化的结合拓展天然酶反应性的巨大潜力。
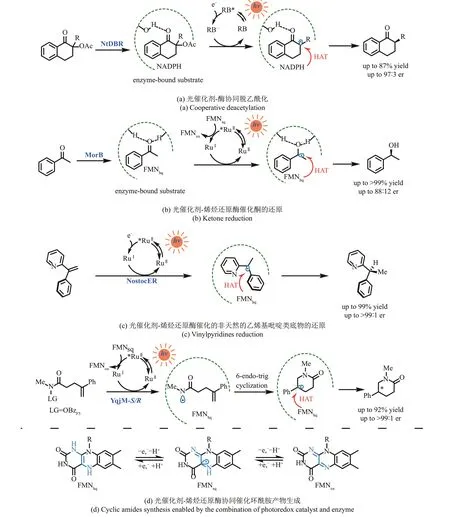
图5 光催化剂选择性还原酶活化的底物以完成非天然转化NtDBR-来自烟草的烟酰胺依赖性双键还原酶DBR;RB-孟加拉玫瑰红;MorB-来自普氏疟原虫的吗啡酮还原酶;RuⅡ-Ru(bpy);NostocER-来自点型念珠蓝细菌的烯烃还原酶; YqjM‑S/R-来自枯草芽孢杆菌的(S/R选择性)烯烃还原酶;OBzF5-五氟苯甲酰氧基Fig. 5 Selected reductions of the enzyme activated substrates by photocatalysts to achieve unnatural transformationsRB-Rose Bengal; MorB-morphinone reductase from P. putida; RuⅡ-Ru(bpy); NtDBR-Double bond reductase from Nicotiana tabacum;NostocER-Ene‑Reductase from N. punctiforme; YqjM‑S/R-Ene‑Reductase from Bacillus subtili; OBzF5-perfluorobenzoyloxy
黄素依赖性烯烃还原酶是催化烯烃不对称还原的高选择性酶催化剂,然而该酶的天然底物仅限于有吸电子取代基(如醛基、羧基、酯基、酮基、腈基和硝基等)的烯烃。2019 年Hyster 课题组[59]利用Ru(bpy)3Cl2在光照下选择性单电子还原与烯烃还原酶结合的酮类底物,通过酮基自由基(ketyl radical)和FMNhq之间的HAT,实现烯烃还原酶对非天然的酮类底物的对映选择性还原[图5(b)]。之后在2020 年,Hyster 等[60]利用类似的策略实现了乙烯基吡啶类底物的对映选择性还原[图5(c)],进一步证明了光酶协同催化对于开发天然酶新功能的重要作用。
Hyster 组[61]近期的一个相关工作描述了通过在蛋白质活性位点内生成氮自由基,进而实现不对称氢胺化反应的例子[图5(d)]。首先,外源光催化剂Ru(bpy)3Cl2被激发后产生长寿命三重态激发态络合物*RuⅡ,其被酶中的辅因子FMNsq还原为RuⅠ,后者将电子转移给与酶结合的肟酯底物,脱去苯甲酸阴离子后,生成氮中心自由基。生成的氮自由基与分子内(或另一分子)的烯烃加成,得到的前手性碳自由基中间体与还原态辅因子FMNhq发生立体选择性的HAT过程,最终生成对映体富集的环化产物。该文报道了5‑exo、6‑endo、7‑endo和8‑endo以及分子间氢胺化产物,并通过定向进化得到了两种对映选择性互补的酶催化剂。
2.4 光生反应中间体参与后续酶促反应
2018 年Castagnolo 等[62]报道了将光催化硫代Michael 加成与生物催化酮还原相结合,实现1,3‑巯基烷醇对映选择性合成的光生物催化级联反应。2019 年,何延红、官智等报道的三联吡啶钌和脂肪酶[63]的结合体系,实现了2,2‑二取代吲哚‑3‑酮的直接不对称合成[图6(a)][64]。首先,2‑苯基吲哚被激发态的光敏剂*RuⅡ单电子氧化为自由基阳离子中间体;而经历单电子转移得到的RuⅠ被O2氧化回基态RuⅡ,以完成光氧化还原催化循环。在此过程中O2被还原为超氧自由基阴离子O·-2,O·-2与吲哚自由基阳离子经过加成、分子内质子转移、脱水过程后形成亚胺酮中间体。最后,小麦胚芽脂肪酶(wheat germ lipase, WGL)的催化三联体[Asp(or Glu)‑His‑Ser]质子化亚胺酮中间体;与此同时,丙酮在酶活性口袋中形成烯醇化阴离子;烯醇化阴离子进攻亚胺离子中间体以生成最终的对映体富集产物。

图6 光生反应中间体参与后续酶促反应Fig. 6 Photogenerated intermediates take part in subsequent enzyme‑catalyzed reactions
H2O2是多种过氧化物酶与过氧合酶的共底物,能够直接参与酶催化的氧化还原反应,避免漫长复杂的电子传递链,提高反应效率[65‑66]。但是高浓度的H2O2或者使用葡萄糖/葡萄糖氧化酶生成H2O时产生的副产物改变体系pH 值[13],均可能引起生物催化剂的失活。Frank Hollmann 等[27]利用无机光催化剂金‑二氧化钛(Au‑TiO2),将甲醇和氧气转化为CO2和H2O2,为过氧合酶rAaeUPO 原位提供H2O2,克服了该酶在高浓度H2O2存在下稳健性差的缺陷,使得酶在催化过程中保持高度活性和稳定性。通过使用该策略,rAaeUPO 将乙苯立体选择性羟化为R‑1‑苯乙醇,并实现了高对映体选择性(>98% ee)和优异的催化转化率(TTN>71 000)。类似地,Hollmann 课题组[67]还开发了以水为最终牺牲电子供体的Au‑TiO2‑H2O2‑rAaeUPO 耦合系统,实现了可见光驱动的C-H 对映选择性氧化。进一步,张武元、Hollmann等[68]在2020年利用光引发的蒽醌‑2‑磺酸钠催化过氧化氢的原位生成,与卤素过氧化物酶(或过氧合酶)协同,实现了多种底物的卤化和羟基化转化[图6(b)]。
3 光激发已知酶实现新转化
上述光催化剂‑酶的协同或串联反应,需要外加光催化剂以完成光激发过程,与酶催化的非光过程是相对独立的。除此之外,能够直接被光激发的一些酶或酶‑底物络合物将在本节重点介绍。目前已经发现的4类天然光酶催化中:光合系统是地球上最重要也最早出现的光酶过程,由光系统Ⅰ和光系统Ⅱ组成,是自然界有机物生产的基础[31‑32];光裂合酶作为一种DNA 修复酶,可以利用蓝光催化光解环丁烷嘧啶二聚体(紫外线照射导致邻近的胸腺嘧啶或胞嘧啶二聚),修复受损DNA[33];原叶绿素酸酯氧化还原酶是一种依赖NADPH的光促酶,通过将原叶绿素还原为叶绿素a,参与叶绿素的合成[34];以及从变异小球藻(Chlorella variabilis)中发现的FAD 依赖的脂肪酸光脱羧酶CvFAP[35‑36]。前三类发现较早,酶的底物特异性也很高,难以用于开发新反应。本部分重点介绍CvFAP 的光催化新转化以及光激发自然界中的非光酶而产生的非天然光反应。
3.1 CvFAP
2017 年, Beisson 及其同事首次报道了CvFAP,证明其可以在可见光激发下,催化一系列中长链(C12~C18)游离脂肪酸脱羧生成相应的烷烃或烯烃[35],之后又对其机理进行了更详细的研究[图7(a)][36]。蓝光激发生成FAD*与脂肪酸阴离子发生迅速的单电子转移过程,随后CO2脱去,留下烷基自由基和FAD 自由基阴离子;之后从FAD 自由基阴离子到烷基自由基发生反向电子转移(back electron transfer, BET),然后通过精氨酸给质子生成产物烷烃,并留下红移态的FADRS。脱去的CO2大约有75%形成碳酸氢盐,用于随后的FAD再生和精氨酸质子化,以完成整个催化循环。

图7 CvFAP的催化机理及其应用实例Arg-精氨酸;FAD-核黄素腺嘌呤二核苷酸;FADRS-红移态的核黄素腺嘌呤二核苷酸;Rac-外消旋Fig. 7 Photobiocatalytic mechanism of CvFAP and the application exampleArg-Arginine; FAD-flavin adenine dinucleotide; FADRS-red‑shifted oxidized flavin; Rac-racemic
自发现CvFAP 以来,研究者们通过酶工程等策略,实现了更多种类的光酶转化。Hollmann等[69]设计了一种诱饵分子接入酶的活性口袋,以实现短链脂肪酸的更有效脱羧,使CvFAP 的底物范围突破长链脂肪酸的限制,实现H2、甲烷、烯烃等燃气的制备。在另一项研究中,吴起等[70]根据晶体结构和分子动力学模拟,通过位点特异性突变在关键位点引入大位阻氨基酸,修饰底物结合通道,使其变得更窄,从而通过疏水相互作用更好地稳定其中一个构型的底物。基于此,他们开发出了CvFAP 的突变体G462Y,能够选择性地催化多种S‑构型的α‑氨基酸和α‑羟基酸的光脱羧反应[图7(b)],以最高达99% ee的对映选择性获得未反应的R‑产物,即实现消旋的α‑官能化羧酸底物的动力学拆分。这项工作是通过酶工程实现CvFAP 不对称催化的第一个例子,同时作者所采用的理性设计[71]的思路可以大大减少酶工程面临的筛选工作的困难。此外,吴起、徐鉴等还报道了将CvFAP 通过酶工程化用于光脱羧氘化[72]和反式脂肪酸的选择性光脱羧[73]的研究,进一步拓展了CvFAP 的催化功能。2022 年,张武元课题组[74]报道了利用CvFAP 将环氧脂肪酸转化成脂肪环氧化物的反应,使用所得脂肪环氧化物进行后续转化,可以生产高附加值脂肪醇、醚和二醇等增值化合物。而偶合脂肪酶、过氧化物酶和光脱羧酶的三酶级联系统,还能以甘油三酯为原料获得脂肪环氧化物。
Scrutton 等[75]利用大肠杆菌和卤单胞菌等底盘表达CvFAP 的突变体,利用生物质或工业废物的脂肪酸生物制造燃料丙烷/丁烷,描绘了一幅规模化绿色生产气体燃料的生物工厂蓝图,有助于为现有化石能源供应提供经济、可持续、安全和清洁的替代品(图8)。而Kabisch 等[76]通过在产油酵母中异源表达CvFAP,整合了甘油转化为脂肪酸和脂质的代谢通路和CvFAP 催化脱羧的新反应,实现了可再生资源到生物燃料的生物制造。此外,吴起、徐鉴等[77]最近还开发了一种光化学‑酶多步组合策略:串联了脂肪酶催化的甘油酯水解、CvFAP 催化的油酸脱羧、长链烯烃的光催化氧化裂解以及CvFAP 突变体催化的中短链脂肪酸的最终脱羧,实现了从三油酸甘油酯到短链烷烃的生物燃料制造。这些工作初步展示出光微生物细胞工厂的巨大应用前景。
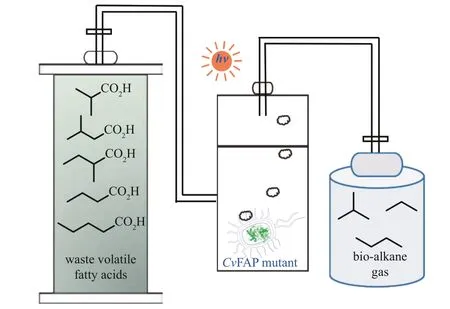
图8 整合了CvFAP突变体的光微生物细胞工厂在生产生物燃料中的应用Fig. 8 Light‑driven microbial cell factories integrating CvFAP mutant for biofuel production
3.2 烟酰胺依赖酶
除了上述天然光酶外,人们陆续发现自然界存在的其他氧化还原酶也可以被光激发,从而诱导酶的混杂反应性。含有烟酰胺和黄素的酶如酮还原酶、烯烃还原酶等在自然界中普遍存在,参与生物体中非光驱动的双电子氧化还原代谢过程[78‑79]。这些酶的辅因子可以通过与非天然底物形成电子供体‑受体复合物(electron donor‑acceptor complex, EDA 复合物)[80‑81],接着通过可见光激发诱发SET 生成自由基物种,实现光诱导的酶的非天然反应。EDA 复合物产生于电子受体和供体之间的前线轨道(HOMO/LUMO)的电子耦合,并可吸收可见光以引发SET,随后生成自由基离子对,通过在底物中添加离去基团以促进该过程向生成自由基的方向不可逆地进行。
2016 年,Hyster 等[82]报道了第一个使用光激发天然非光酶,实现非天然单分子反应的例子(图9)。作者使用的酮还原酶是一类NADPH 依赖的氧化还原酶,通常催化羰基的还原,而在该案例中,卤代内酯底物与酶口袋中的NADPH 形成EDA 复合物,并通过光激发的单电子转移以及随后的氢原子转移,实现了卤代内酯的不对称自由基脱卤化反应。最近,吴起、徐鉴等[83]报道了光诱导黄素依赖的环己酮单加氧酶(CHMO)的还原脱卤混杂反应性,可以将α-卤代‑α‑氟代酮对映选择性地还原为手性α‑氟代酮。通过机理实验和模拟计算揭示了其电子转移/质子转移机制(ET/PT),与以往报道的天然还原酶的光诱导脱卤反应不同,该文提出脱卤后生成的自由基中间体不是通过HAT 生产最终产物,而是经历ET/PT 过程分别从辅酶和溶剂中获得电子和质子,从而生成最终产物。

图9 光诱导醇脱氢酶实现卤代内酯的脱卤化LKADH-来自克菲里乳杆菌的短链脱氢酶;RasDH-来自雷氏菌属的短链脱氢酶Fig. 9 Dehalogenation of halogenated lactones by light‑induced alcohol dehydrogenaseLKADH-short‑chain dehydrogenase from Lactobacillus kefiri;RasDH-short‑chain dehydrogenase from Ralstonia species
2022 年,赵惠民、王斌举等报道了光诱导的酮还原酶催化的分子间自由基共轭加成反应(图10)[84]。作者借鉴了聚焦理性迭代位点特异性突变策略(focused rational iterative site‑specific mutagenesis, FRISM)[71,85]对商品化的酮还原酶P2‑D12 进行了半理性改造,筛选出具有高产率和对映选择性的酶突变体。通过理论计算和机理研究,作者对该反应的机理和立体选择性的起源进行了较全面的解释:与酮还原酶结合的NADPH 和相邻的自由基前体邻苯二甲酰亚胺活化的酯通过π‑π堆叠相互作用形成EDA 复合物,在光激发下经历SET 过程,后续经历N-O 键断裂并脱羧产生烷基自由基。富电子的烷基自由基对有缺电子基团的α,α‑二取代末端烯烃进行共轭加成,生成前手性自由基中间体,最后通过立体选择性的HAT 步骤,实现α‑羰基立体中心的构建。
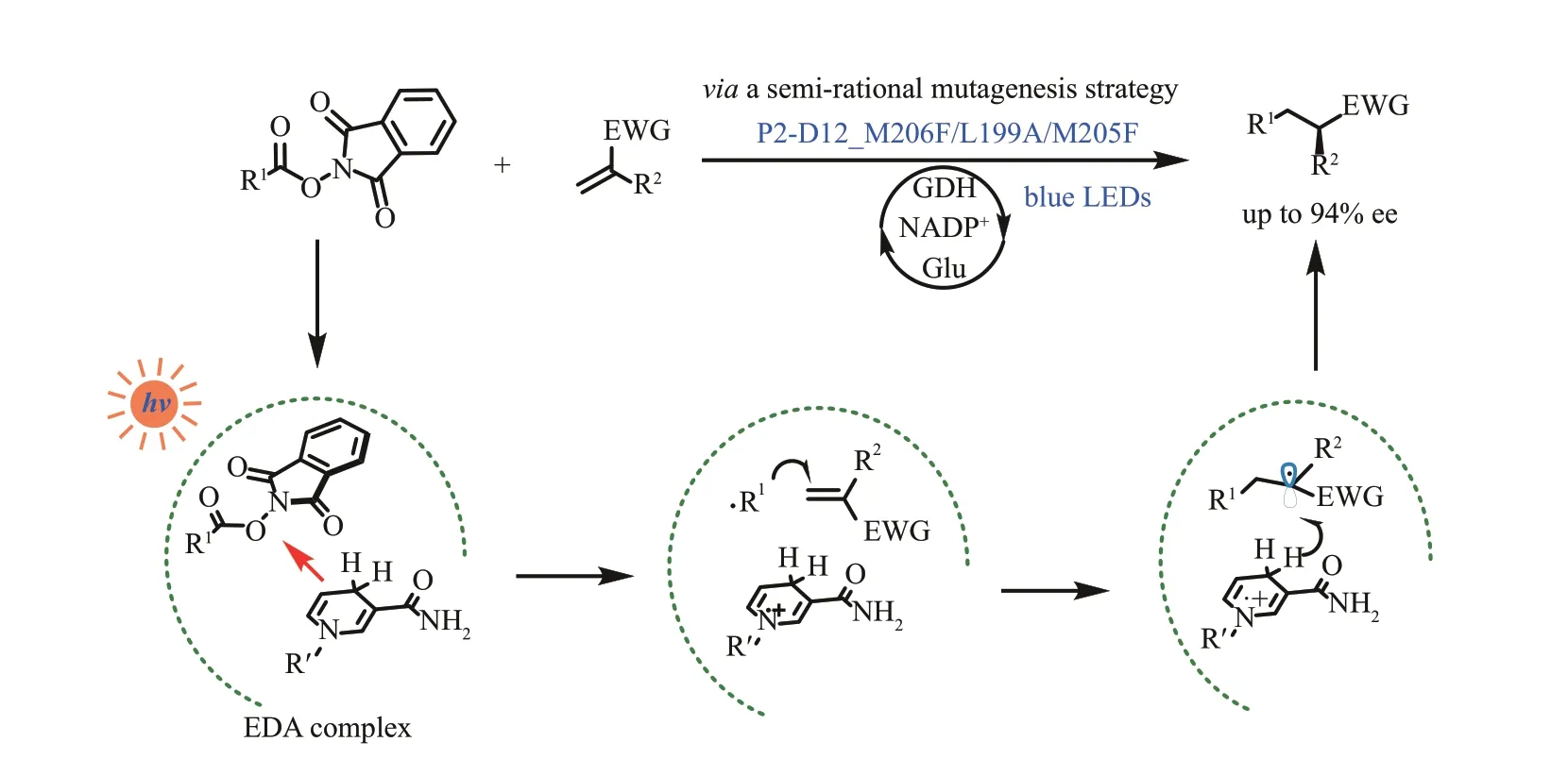
图10 光诱导酮还原酶催化的对映选择性分子间自由基共轭加成反应Fig. 10 Enantioselective intermolecular radical conjugate addition by light‑induced ketoreductase
3.3 黄素依赖的烯烃还原酶
2019 年,Hyster 课题组[86‑87]报道了一例光激发烯烃还原酶实现的分子内自由基环化反应,构建出对映体富集的五元、六元、七元和八元内酰胺[图11(a)]。机理研究表明,通过光激发底物与酶活性位点内的还原态黄素辅因子FMNhq之间形成的EDA 复合物,发生电子转移,底物脱去离去基团氯离子后生成碳自由基,对分子内的烯烃基团进行exo‑加成或endo‑加成,得到β‑或γ‑前手性立体中心,再通过FMNsq对后者进行氢原子转移,得到最终的环化产物。立体化学控制是由酶口袋对于自由基加成方向以及后续HAT 步骤的限制实现的。这个案例有力证明了利用可见光激发口袋内的底物‑辅因子络合物,能开发出基态无法进行的自由基化学,实现天然酶的新催化活性。2022 年,Hyster 等[87]利用类似的策略实现了自由基分子内环化及烯丙基化反应。
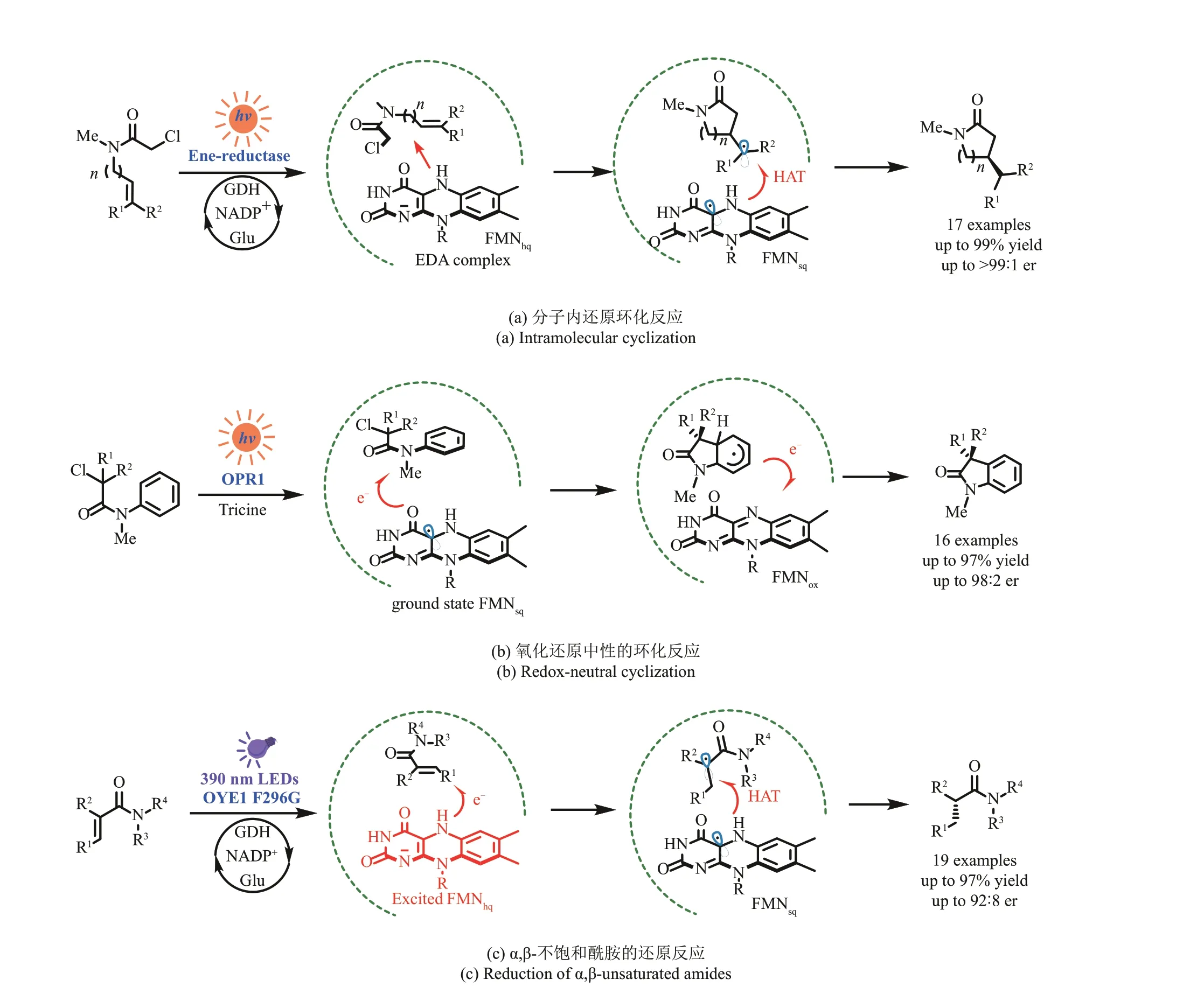
图11 光诱导烯烃还原酶实现的非天然转化OPR1-12‑氧代二甲酸还原酶;OYE1 F298G-老黄酶1的F298G突变体Fig. 11 Light‑induced ene‑reductase catalyzed unnatural transformationsOPR1-12‑oxophytodienoate reductase; OYE1 F298G-mutants F298G of OYE1
2019年,Hyster课题组报道了一例光激发黄素依赖的烯烃还原酶催化的氧化还原中性的不对称自由基环化反应[图11(b)][88]。与之前的例子不同在于,这个例子中没有使用NADPH 辅因子再生系统,而是用了还原性的三(羟甲基)甲基甘氨酸缓冲体系(tricine buffer)。在光照下,tricine 将一个电子转移给FMN,生成FMNsq,基态的FMNsq将底物单电子还原,脱去离去基团氯离子后生成前手性的三级烷基自由基,后续的前手性自由基对苯环的加成过程受酶口袋环境的调控。然后进一步单电子转移和去质子化,完成重新芳构化得到最终的环化产物。
2020 年,Hyster 等[89]报道了一例机理与以往通过EDA 复合物介导的光酶反应不同的案例[图11(c)]。作者使用紫外光直接激发烯烃还原酶口袋内的还原态FMNhq,使其能够还原天然生物催化中低反应性的酰胺取代的烯烃,再经过质子化和酶控制的HAT 过程,实现非天然的α,β‑不饱和酰胺的对映选择性还原。该策略通过直接激发酶分子中的辅因子,拓宽了天然酶促烯烃还原反应的底物范围。
2020 年,赵惠民等[90]报道了第一例光诱导酶催化的非天然两分子交叉偶联反应[图12(a)]。从烯烃与α‑卤代羰基化合物出发,通过可见光诱导的烯烃还原酶催化的自由基氢烷基化途径进行分子间偶联,实现γ‑手性羰基化合物的构建。在优化条件下,老黄酶OYE1可以以优异的产率和对映异构体选择性合成多种含有γ‑立体中心的羰基化合物。在不加烯烃底物的情况下能够观测到大量脱卤副产物,这表明FMNhq和α‑卤代羰基化合物在酶口袋内形成的EDA 络合物,在光激发条件下足以引发单电子转移的过程。2021 年,Hyster 课题组[91]报道了一例相似的分子间氢烷基化反应。作者通过瞬态吸收光谱等机理实验,提出该反应中烯烃还原酶、酶口袋中的FMNhq、卤代酰胺以及另一底物α‑甲基苯乙烯生成的四元复合物,是可见光激发的物种。其中α‑甲基苯乙烯的存在可以提高FMNhq电子转移到氯代酰胺底物的量子产率。随后,Hyster课题组利用烟酰胺依赖型环己烯酮还原酶(NCR)的变体,报道了一例基于基态单电子转移机制完成的烯烃氢烷基化反应[92]。
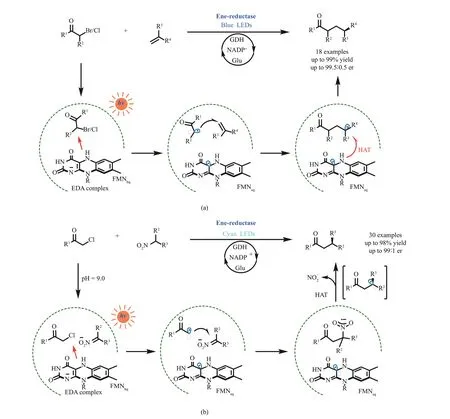
图12 光激发烯烃还原酶实现的分子间烯烃氢烷基化反应(a)和Csp3-Csp3亲电交叉偶联反应(b)Fig. 12 Photoactivated ene‑reductases enabled intermolecular reductive coupling couplings for alkene hydroalkylations (a) and Csp3-Csp3 bond formations (b)
2022 年,Hyster 课题组[93]报道了一例光酶催化的卤代烷烃和硝基烷烃的不对称Csp3-Csp3亲电交叉偶联反应[图12(b)]。α‑卤代羰基化合物和烯烃还原酶口袋中的FMNhq形成EDA 复合物,在光激发后优先于硝基烷烃被化学选择性地单电子还原,进一步脱卤形成烷基自由基。关键的C-C键形成步骤是硝基烷烃去质子化原位生成的碳负中心捕获缺电子的α‑羰基碳自由基,然后经过亚硝酸根离子脱去步骤,形成前手性碳自由基中间体,最后经过酶控制的HAT 实现立体中心的构建。作者筛选出两种烯烃还原酶CsER 和GluER‑T36A,分别催化S‑对映体和R-对映体富集产物的生成。值得注意的是,与之前常用的偏中性的缓冲体系不同,该反应的缓冲液pH调到了9.0,有利于硝基烷烃的去质子化。
4 人工光酶
人工光敏基团和/或人工催化基团可以与蛋白骨架结合,以赋予酶蛋白全新的反应性;反过来,酶骨架可以稳定在水相中的人工辅因子,并可能为反应提供受限环境或氢键作用,从而促进反应发生或实现选择性,这是人工光酶的优势。人工光酶的构建目前主要有两种策略:①通过将光敏基团与(非天然)氨基酸共价连接构建,利用光敏基团促进反应底物的直接SET过程;②在蛋白质骨架内,通过密码子扩增技术引入非天然光敏基团,并通过光引发的电子或能量转移来促进反应的发生。
4.1 共价交联酶和光催化剂
2015 年,Lewis 等[94]通过叠氮化物‑炔环加成的点击化学策略,将9‑甲磺基‑10‑甲基吖啶(9‑mesityl‑10‑methylacridinium, Acr+‑Mes)光敏基团共价连接到脯氨酰寡肽酶(POP)内的非天然氨基酸残基上,构建了一种人工光酶,用于硫茴香醚的氧化生成亚砜[图13(a)]。为了更好地引入人工辅因子,研究者在该酶的活性口袋内引入4‑叠氮基‑L‑苯丙氨酸,同时将附近4 个氨基酸残基突变为丙氨酸,从而开发一个可以容纳大空间位阻的人工辅因子的口袋。机理研究表明,该反应通过酶活性位点内硫醚底物的电子攫取开始,蛋白骨架延长了吖啶辅因子的荧光寿命。然而对照实验结果显示,虽然光敏剂成功修饰到生物催化剂上,但与单独的Acr+‑Mes 相比,人工光酶催化的产率并无提高,甚至有的更低。后来他们又将RuⅡ多吡啶络合物引入POP 蛋白骨架,通过修饰吡啶上的取代基以及筛选不同的引入位点,得到了能够催化二烯酮的分子内还原环化,以及肉桂酰基咪唑和4‑甲氧基苯乙烯之间的[2+2]光环加成反应的人工光酶[图13(b)][95]。该酶相比于游离的Ru(bpy)有更高的产率和反应速率,但是无法控制反应的立体化学选择性。
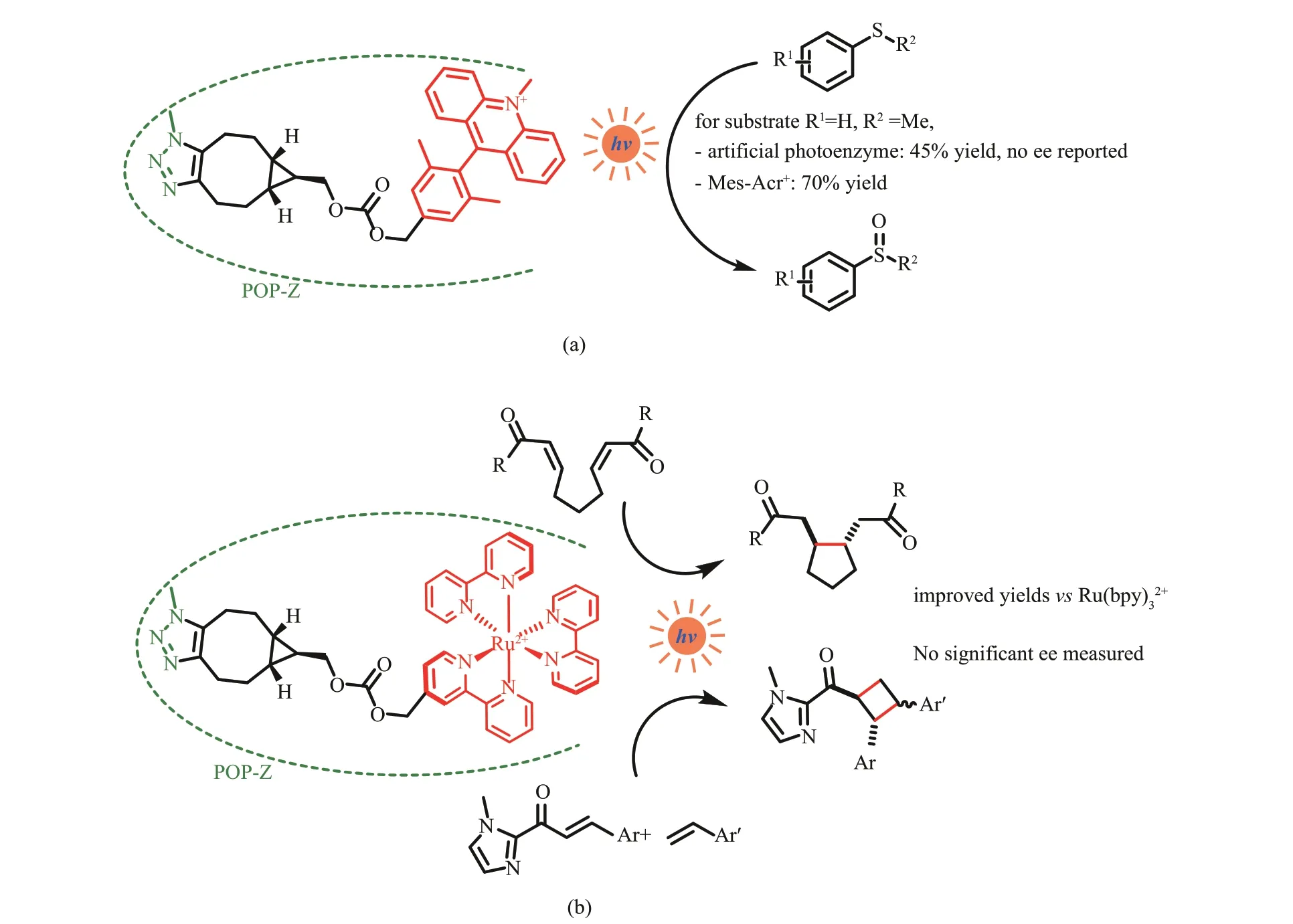
图13 通过点击化学将吖啶类光敏剂(a)、三联吡啶钌(b)引入蛋白POP‑Z-引入非天然4‑叠氮基‑L‑苯丙氨酸脯氨酰寡肽酶Fig. 13 Introduction of acridine photosensitizer (a) and tris(2,2'‑bipyridyl) rutheniumⅡ (b) into protein by clicking chemistryPOP‑Z-p‑azido‑L‑phenylalanine (Z) incorporated prolyl oligopeptidase (POP)
2018 年,Cheruzel 等[96]通过蛋白表面的半胱氨酸残基与碘代乙酰胺衍生物的共价交联反应,构建了一种P450 BM3 和钌(Ⅱ)光催化剂的杂合体。通过可见光激发引发单电子转移,提供P450催化所必需的还原当量,从而实现了光引发的P450 催化的天然反应--C-H 键羟基化。2020年,Brustad 等[97]通过碘代乙酰胺手柄将2 种Acr+‑Mes 辅因子与3 类稳定且具有大空腔的蛋白质骨架中的半胱氨酸结合,开发出12 种人工光酶,用于硫茴香醚的氧化(图14)。作者发现这些人工光酶的催化性能取决于多种因素,包括Mes‑Acr+光敏辅因子、蛋白质骨架、辅因子固定的位置以及不同底物等,但由于是在酶表面的凹槽处引入的非天然光敏基团,因此亚砜产物并没有任何立体选择性,同时反应效率与未结合的Mes‑Acr+相比并没有显著优势。

图14 通过半胱氨酸残基与碘代乙酰胺衍生物共价交联引入光敏剂构建不同人工光酶Fig. 14 Construction of different artificial photoenzymes by introducing photosensitizers through covalent cross‑linking of cysteine residues with iodoacetamide derivatives
2022 年,Schlau‑Cohen 等[98]通过将商品化的红藻光合作用捕光蛋白R‑藻红蛋白(R‑phycoerythrin, R‑PE)与多个[Ru(bpy)3]2+光催化剂通过酰胺键连接,形成新的生物杂化光催化剂,能够利用630 nm 的红光,催化硫醇与烯烃的偶联以及半胱氨酸的脱硫反应等(图15)。光谱学等机理研究表明,该杂化光催化剂中,红光的能量也能够从RPE 蛋白有效转移至光催化剂,以增强化学反应性,提高产率。此外,该生物杂化催化剂虽然是一种均相催化剂,却能够通过超滤管离心过滤实现重复利用。该策略为开发独特的生物杂化光催化剂提供了新的思路。之后,Hyster、Schlau‑Cohen 等[99]共价连接了烯烃还原酶与经典的有机染料光催化剂,获得了能够被低能长波长可见光激发的新人工光酶,能够有效催化分子内的自由基环化反应。
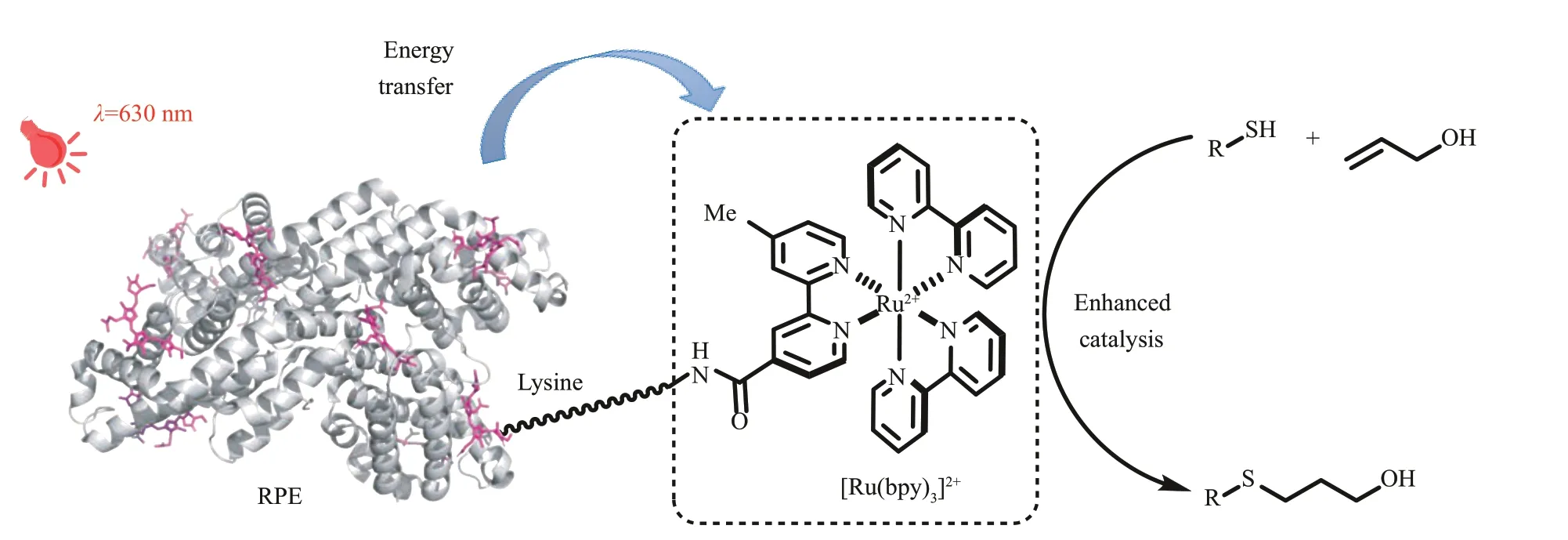
图15 通过光合吸光蛋白与光催化剂结合构建吸收低能光的光酶RPE-R‑藻红蛋白,PDB 1EYXFig. 15 Construction of low energy absorption photoenzyme via the combination of photosynthetic light‑harvesting protein and photocatalystRPE-R‑phycoerythrin, PDB 1EYX
4.2 通过基因密码子扩增技术插入非天然光敏氨基酸+共价交联催化活性基团
王江云及其合作者[100]使用基因密码子扩增技术将非天然氨基酸插入荧光蛋白(photosensitizer protein, PSP),得到具有二苯甲酮核心骨架的光敏基团[101‑102],进一步在蛋白表面特定位置,通过半胱氨酸与碘代乙酰胺衍生物共价交联的方式,引入小分子镍络合物,构筑了一类新颖的人工光酶。2018 年,基于光引发的单电子还原和三联吡啶镍络合物,实现了光驱动的二氧化碳还原反应[图16(a)][103]。2020 年王江云、钟芳锐、刘晓红和吴钰周等[104]将二苯甲酮光敏基团与PSP 蛋白表面修饰的二联吡啶镍络合物相结合,基于紫外光引发的能量转移过程和过渡金属催化的交叉偶联,实现了人工光酶催化的卤代芳烃脱卤羟化反应以及C-N 键构建反应[图16(b)]。研究发现,通过精确调控光敏基团与催化基团之间的空间距离,有望提高双催化的协同作用。
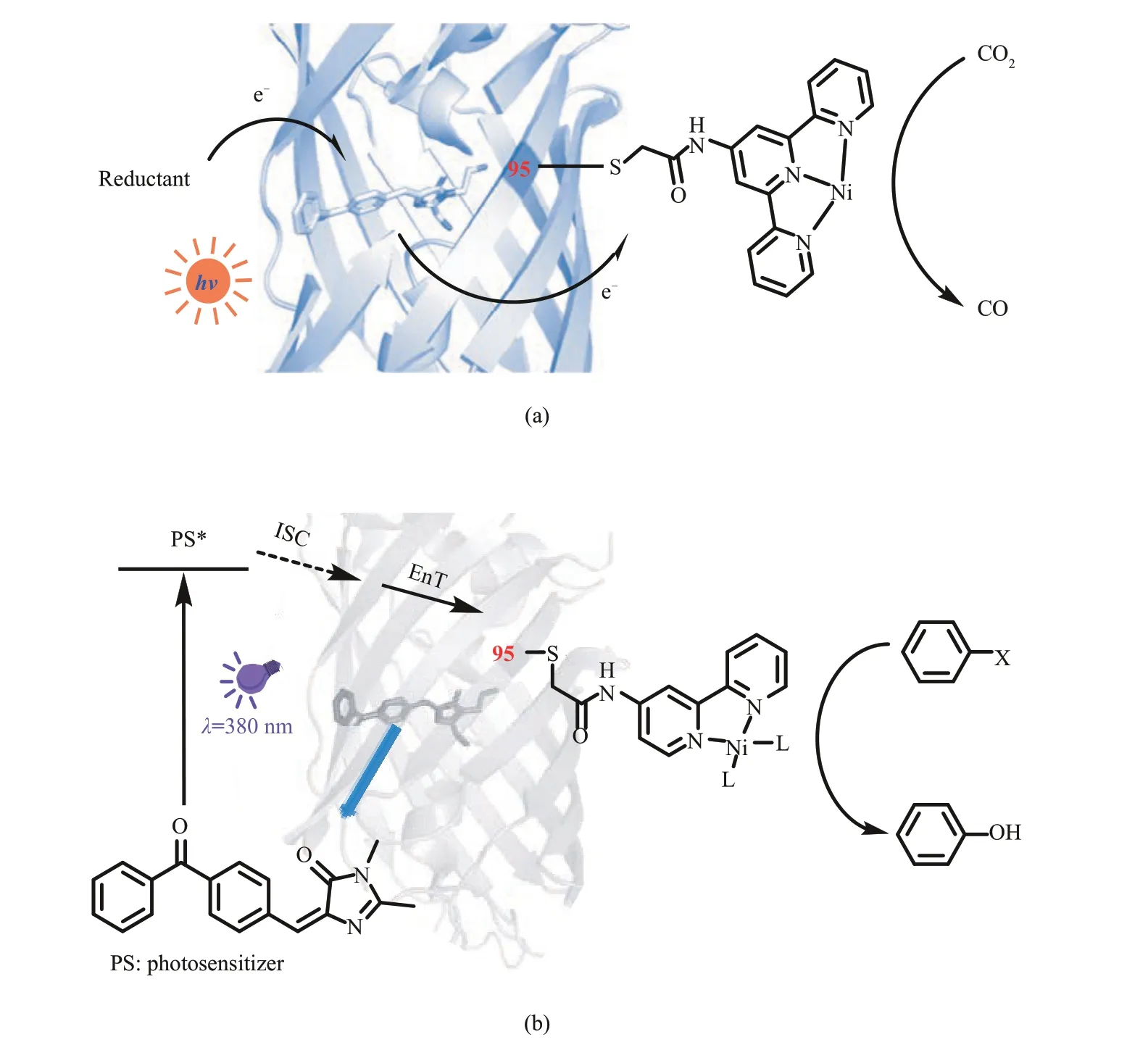
图16 具有二苯甲酮光敏基团的人工光酶催化二氧化碳的还原(a);卤代芳烃的脱卤羟化反应(b)Fig. 16 Artificial photoenzymes with benzophenone photosensitive groups catalyze the reduction of carbon dioxide (a) and the dehalogenation and hydroxylation of aryl halides (b)
4.3 通过基因密码子扩增技术插入非天然光敏氨基酸催化立体化学控制的能量转移反应
2022年,吴钰周、钟芳锐和陈希等[105]报道了一类新颖的人工光酶,能够在紫外光引发下,催化立体选择性的三线态能量转移反应[图17(a)]。作者通过遗传密码子扩增技术,在多药耐药性调节蛋白LmrR[106]骨架中插入二苯甲酮类光敏基团,构建的人工光酶能够通过三线态能量转移机制,催化吲哚衍生物的对映选择性分子内[2+2]环加成去芳构化反应。通过4 次计算机辅助的理性诱变,作者将该酶的结构进行了优化,最终得到了一种高效的人工光酶突变体,有着显著的底物通用性和优异的对映体选择性。
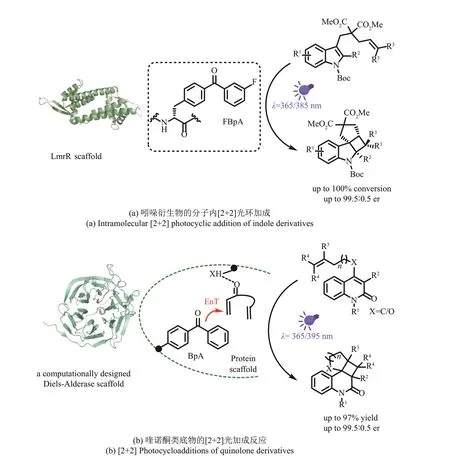
图17 人工光酶实现对映选择性的能量转移反应Fig. 17 Enantioselective energy transfer reactions realized by artificial photoenzymes
同一时间,Green 等[107]报道了相似策略构建的人工光酶,通过三线态能量转移促进对映选择性[2+2]环加成反应[图17(b)]。作者利用遗传密码子扩展技术将含有二苯甲酮光敏基团的非天然氨基酸引入计算机从头设计的一种DA 酶[108]疏水口袋中的173号位,得到能够催化三线态能量转移的人工光酶母本。定向进化改造后得到的人工光酶突变体,可实现最高达99%的转化率和99%的对映异构体过量值。
这两项研究利用遗传密码子扩增技术,将三线态能量转移机制与蛋白质相结合,并通过高通量筛选的酶工程,开发出具有更高反应活性和选择性的人工光酶变体,为开发新一代具有类似于天然酶的效率和特异性的可进化光酶催化剂建立了框架。
5 展 望
尽管光酶催化刚刚兴起,但是该策略已经在合成科学领域展现出巨大的应用前景。表1总结了目前报道的光酶催化反应的主要案例。光催化能够产生高活性的中间体,而生物催化在控制立体选择性方面具有独特的优势,光酶催化很好地综合了两者的优势,能够完成众多非天然化学转化。光与酶的协同体现在多个方面:光催化可以实现生物催化中辅因子的循环,达到节约成本或者更高效的电子传递等目的;具有光活性的辅因子的利用以及辅因子‑底物EDA 复合物形成的策略,进一步拓宽了生物催化的化学转化边界,也实现了部分传统化学催化剂无法完成的不对称转化;酶可以通过定向进化来进行工程化改造,内部的氢键网络及其他相互作用力可以稳定活泼的开壳中间体、控制反应的立体化学过程;为了探索更广阔的应用前景,人工光酶逐渐出现,并进一步突破了现有酶的应用范围,使得人们可以根据反应需求设计新的生物催化剂。
过去的几十年,我们见证了现代可见光催化合成的蓬勃发展和应用,以及蛋白质工程和合成生物学新技术的不断涌现。因此,光与酶的结合方兴未艾,具有巨大的机遇,同时也面临着众多挑战:
(1)当前可被可见光激发的酶的种类比较少,多局限于黄素和烟酰胺依赖的氧化还原酶,这也导致了光酶催化的非天然转化面临机理途径较单一(多是基于EDA复合物引发的净还原体系)、依赖于活泼的自由基前体等问题。因此,急需创制可被可见光激发的新生物催化剂,比如具有多种价态变化的金属酶与人工光酶等,进而开发更多的光酶协同机理路径,实现更多更广的非天然底物到高附加值光学纯手性产品的合成。
(2)人工光酶用于催化非天然的不对称转化刚刚开始,亟待创制更多种类丰富、催化性能强大、构建方法简洁高效、光量子效率更高的新型人工光酶。除了自上而下的酶工程,从头设计全新的人工光酶,通过在蛋白质内产生全新的催化中心和/或光敏中心,可以为人工光酶的创制提供更快更有效的解决方案,从而为新的不对称光酶催化转化提供更多机会。
(3)不同于经典酶,光酶通常面临氧气不耐受的问题,即氧气的存在可能会猝灭光诱导的反应中间体或产生使蛋白质失活的活性氧物种。此外光酶催化的非天然新反应,往往还存在着立体选择性不高、活性差等问题,而传统的酶进化方法只对蛋白局部优化,面临活性和稳定性不可兼得等问题。因此,综合利用机器学习和高通量自动化生物制造平台,建立光酶的多目标协同的、高效标准化的定向进化平台,进而实现其功能全面提升,是光酶催化合成领域待研究的方向之一。
(4)目前的光生物催化体系多数仅限于实验室规模研究,更多有价值的新反应无法实现工业化生产。这是由于光的穿透性差以及光酶催化剂用量较大等问题,阻碍了光酶催化合成的放大。未来可用的解决方案包括引入内部照明或者连续流光反应装置进行生产、进一步改造蛋白提高光酶的TOF、固定化光酶、发展高效光微生物细胞工厂等。
通过交叉整合生物合成与化学合成,我们相信人们会发现更多有趣、有用的新光酶催化转化,并且开发出用于规模化生物制造的光酶催化工艺与光微生物细胞工厂,逐步推进“绿色光生物制造”时代的到来。
