溶血性贫血导致肝紫癜病和继发性含铁血黄素沉积症1例及文献复习*
2023-09-12安薪宇胡灵溪刘百成任伟光王荣琦南月敏
李 妹,安薪宇,胡灵溪,刘百成,任伟光,张 莹,王荣琦,南月敏
肝紫癜病(peliosis hepatis,PH)是一种较少见的肝脏良性病变,以肝实质内多发大小不等的充满血液的囊腔为主要表现,也可同时累及肺和网状内皮系统器官,如脾脏、淋巴结和骨髓等[1]。该病病因暂不明确,且缺乏特异性的临床表现,易被误诊。现将我科收治的1例溶血性贫血导致的PH和继发性含铁血黄素沉积症患者的临床资料报道如下,并结合相关文献,就其病因、发病机制、诊断方法和治疗手段予以讨论,旨在进一步提高临床医师对血液系统疾病所致肝损伤的认识,以减少误诊和漏诊。
1 病例资料
患者男性,54岁。主因间断眼黄、尿黄6年,加重1月于2022年1月18日入院。缘于6年前无明显诱因下出现眼黄、尿黄,淡茶水样,就诊于当地医院,诊断为“肝炎”,血清HBV标记物和HBV DNA结果不详,间断口服中药治疗。半年前,患者无明显诱因出现眼黄、尿黄,行相关化验检查,诊断为“肝硬化、脾肿大、脾功能亢进症、乙型肝炎、丙型肝炎”,行脾切除术治疗。术后,间断复查肝功能和血常规,均未见明显异常。3月前,无明显诱因再次出现眼黄、尿黄,伴乏力,就诊于我院门诊,化验血常规:WBC 10.0×109/L、HGB 80.3 g/L、PLT 116.0×109/L;肝功能:AST 16.0 U/L,ALT 23.0 U/L,TBIL 112.3 μmol/L,DBIL 29.8 μmol/L。肝胆脾超声检查提示肝硬化声像图表现。住我院血液科,行骨髓穿刺术,提示红系比例明显增高,考虑增生性贫血骨髓象。血清直接抗人球蛋白试验阳性。考虑自身免疫性溶血性贫血(autoimmune hemolytic anemia,AIHA)。患者未接受进一步治疗,自动出院。1月前,上述症状加重,为求进一步治疗,入住我科。自发病以来,精神、饮食和睡眠尚可,尿色黄,粪便正常,无粘液脓血便或黑便。体质量下降不明显。患者既往体健,无结核等传染病史。有输血史。无食物、药物过敏史。查体:T 36.5℃,P 79次/分, R 20次/分,Bp 121/97 mmHg。慢性肝病面容,无肝掌、蜘蛛痣,全身皮肤、粘膜黄染,结膜苍白,巩膜中度黄染,心肺听诊未见异常,腹平软,左肋下缘可见长约25 cm手术瘢痕,腹部无压痛、反跳痛,肝肋下未触及,肝区无叩痛,腹水征阴性。双下肢无水肿。辅助检查:血常规:WBC 5.9×109/L、NEUT 3.1×109/L、RBC 2.8×1012/L、HGB 68.8 g/L、PLT 529.0×109/L、NRBC 1.3×109/L、NRBC% 12.0%;血清ALB 39.7 g/L、ALT 21.0 U/L、AST 28.0 U/L、TBIL 142.3 μmo1/L、DBIL 58.2 μmol/L;PT 13.5 s,PTA 64.2% ,INR 1.3,APTT 32.8 s;血清HBsAg、HBeAb和HBcAb阳性,HBV DNA<20 IU/ml,抗-HCV阳性,HCV RNA<50IU/ml。尿常规提示尿胆原(3+),尿胆红素阴性。血清抗核抗体、抗dsDNA抗体、CD55、CD59、EB病毒、巨细胞病毒、人类免疫缺陷病毒、梅毒血清标志物均阴性。腹部超声提示肝硬化。CT见右肺中叶肺气肿,肝实质密度略高,肝右叶见条状或片状更高密度影。给予护肝退黄,改善循环等对症支持治疗。因以血清间接胆红素升高为主,考虑与血液原发病有关,不除外Gilbert综合征可能。行肝穿刺活检,组织病理学检查提示,肝细胞内可见大量色素颗粒沉积,铁染色证实为铁颗粒,部分小叶中央静脉周围轻度纤维化,肝窦扩张,肝细胞受压,肝板萎缩,部分肝窦内可见淤血,肝细胞核大小不一,可见大核、双核肝细胞;窦周纤维化不明显;汇管区扩大,炎细胞浸润不明显,纤维组织增生,小叶界板大致完整。Cu染色(-),Fe染色(强阳性)。免疫组织化学染色HBsAg(++,胞浆型),HBcAg阴性(图1~3)。病理学诊断: 1,PH; 2,继发性含铁血黄素沉积症;3,慢性乙型肝炎和丙型肝炎重叠感染。在对症支持治疗的基础上,给予甲泼尼龙琥珀酸钠40 mg静脉滴注,1次/d,2天后改为甲泼尼龙片36 mg口服,1次/d,并逐渐减量至20 mg 1次/d。因激素可能激活病毒复制风险,遂加用恩替卡韦分散片0.5 mg口服,1次/d,抗病毒治疗。10天后复查,WBC 10.2×109/L,RBC 3.9×1012/L、HGB 98.5 g/L,PLT 565.0×109/L;血清ALB 41.7 g/L、ALT 49.0 U/L、AST 31.0 U/L、TBIL 81.9 μmol/L、DBIL 34.0 μmol/L。于2022年1月30日出院。院外继续口服甲泼尼龙20 mg /d,恩替卡韦0.5 mg/d,水飞蓟宾胶囊70 mg口服,3次/d,熊去氧胆酸胶囊0.25 g 3次/d。对于PH,因影像学检查未见肝脏有局限性或弥漫性低密度灶,且患者临床症状较轻,未出现发热、上腹痛、腹水等症状,遂暂未予特殊处理。对于继发性含铁血黄素沉积症,也暂观察随访,必要时可加用铁螯合剂。1个月后,于当地医院复查血常规和肝功能,均较前明显好转,遂自行停用甲泼尼龙治疗,现病情平稳。
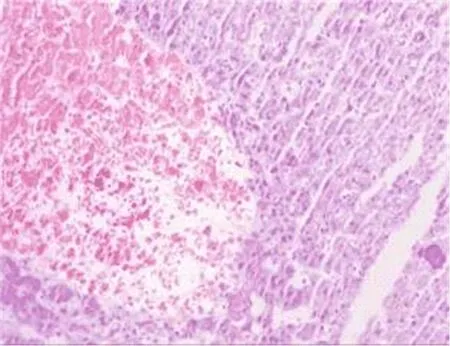
图1 肝组织病理学表现 肝窦扩张,肝细胞受压,肝板萎缩,部分肝窦内可见淤血(HE,200×)

图2 肝组织病理学表现 汇管区扩大,炎细胞浸润不明显,纤维组织增生,小叶界板大致完整(Masson染色,200×)
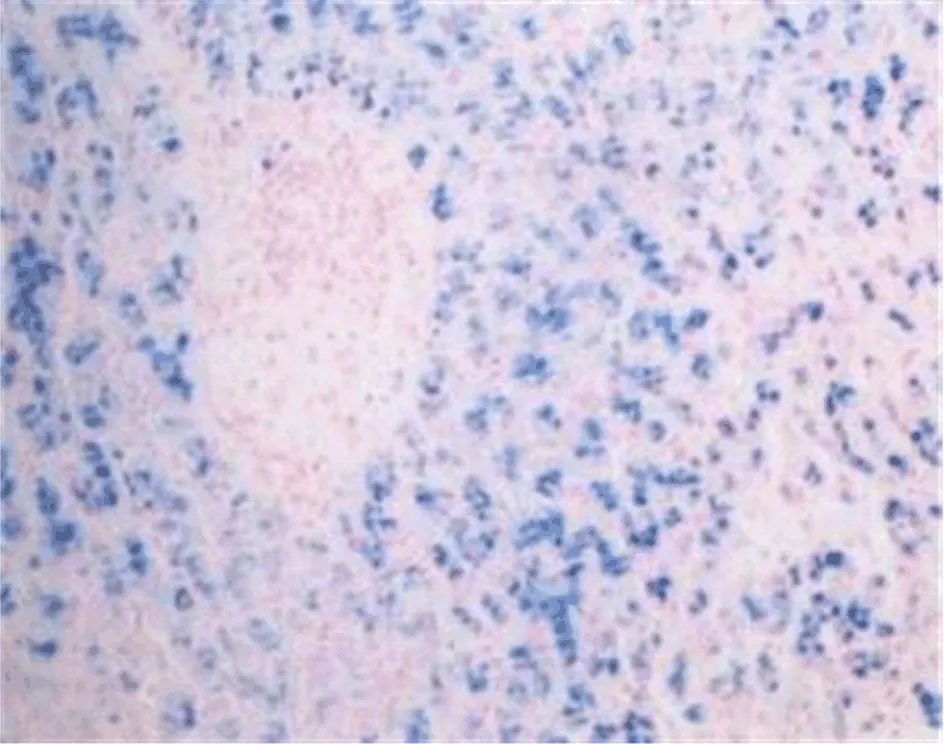
图3 肝组织病理学表现 肝细胞内可见大量色素颗粒沉积,铁染色证实为铁颗粒,铁染色强阳性(Fe染色,200×)
2 讨论
1861年Wanger首次在文献中报道了PH,并于1916年由Schoenlank命名[2]。目前,PH的病因尚不明确,可能与严重的结核病、恶性肿瘤、多发性骨髓瘤和艾滋病等有关,也可能与长期应用类固醇类药物、免疫抑制剂、避孕药和硫唑嘌呤等有关[1,3]。PH的发病机制仍存在争议,大概包括以下几个方面[1,3,4]:1)血管先天畸形;2)肝细胞坏死及蛋白网状支架结构的破坏导致肝窦囊性扩张;3)肝窦与中央静脉连接部受阻使肝窦淤血扩张;4)肝窦屏障被破坏,继发肝窦扩张和狄氏(Disse)间隙扩大,形成充满红细胞的囊腔,阻塞门静脉血流。该病无特异性的临床表现,易将其误诊为肝细胞癌、包虫病等。临床分型为局灶型和弥漫型两种类型。
该病超声检查可提示肝肿大,多发性不均匀低回声区[5]。CT检查多表现为低密度灶,增强扫描动脉期更明显,病变中心可见球形强化影,呈“靶征”;门静脉期病灶强化范围呈“离心性”或“向心性”扩展;实质期病变呈弥漫均匀强化,周边呈低密度影像[6]。MRI多表现为T1病灶呈低信号,T2呈高信号[7]。CT和MRI的表现均取决于病灶大小和出血情况,不同时期血液成分不同导致影像学表现亦有所不同。因影像学诊断缺乏特异性,肝组织活检仍为诊断该病的金标准,但该操作有出血风险,应谨慎进行。
治疗PH的主要原则包括控制诱因、对症治疗、定期随访和预防并发症等。对于病灶<5 cm、无症状的患者可暂不处理,随诊观察;当病灶>5 cm 时,可采用微波、射频消融等技术治疗;若病灶较大且有明显压迫症状或单发的有破裂出血倾向时,可行手术切除;若病变呈弥漫性表现且合并肝功能失代偿,可考虑行原位肝移植或肝移植术。有研究发现[8],溶血性贫血导致肝损伤的原因有两个,一是变形或溶血的红细胞溶解物粘附于肝血管内皮,致内皮细胞受损,形成充满红细胞的囊腔,阻塞门静脉血流,导致PH;二是溶血性贫血致肝窦被破碎的红细胞堵塞,从而使肝窦与中央静脉连接部受阻,肝窦淤血扩张,导致PH。结合该患者的临床症状和辅助检查,考虑其为局灶型PH,可暂不予以特殊处理,故给予控制原发病及护肝退黄等对症支持治疗,随诊观察。
含铁血黄素沉积症又称血色病(hemochromatosis,HC),根据其病因分为原发性和继发性。原发性血色病(hereditary HC,HHC)是一种常染色体隐性遗传病,由位于第6号染色体短臂上的基因发生突变所致。在北欧人群常见[9]。继发性血色病(secondary hemochromatosis,SHC)主要由过量应用铁剂、长期慢性贫血、输注红细胞、病毒或酒精致肝硬化等,造成过量铁沉积所致。放血疗法是治疗该病的主要手段。对于合并慢性溶血性贫血患者,可应用铁螯合剂[9]。根据该患者病例特点,考虑其为继发性血色病,且处于早期阶段,故在积极治疗原发病的基础上,密切观察随访,必要时加用铁螯合剂治疗。
AIHA是由各种原因导致机体免疫功能紊乱,产生抗自身红细胞抗体导致红细胞破坏的一种溶血性贫血[10]。糖皮质激素是首选治疗,有效率在80%以上。若激素治疗4周仍未控制病情,可考虑二线治疗,即行脾切除、利妥昔单抗和细胞毒性免疫抑制剂等[10]。该患者贫血、黄疸、血清间接胆红素升高;直接抗人球蛋白试验阳性;除外系统性红斑狼疮、类风湿性关节炎或巨细胞病毒感染等继发因素,符合AIHA诊断[10]。有研究发现[11],对于AIHA患者,脾切除术后仍口服小剂量激素维持治疗,可大大提高1年病情缓解率。该患者已行脾切除术,但仍有溶血性贫血的表现,故在对症支持治疗的基础上给予激素冲击治疗,可见血红蛋白较前明显升高,后改为口服小剂量激素维持治疗,病情逐渐缓解。
总之,当肝脏疾病与血液系统疾病密切相关时,肝病医师在临床诊疗实践过程中应根据患者肝损伤情况,追根溯源,及早明确诊断,并给予对症处理,才能提高诊疗水平,改善患者预后。
