重症监护室肺部感染患者肠道菌群特征分析*
2023-09-11蒋小艳王浩园范文辉周茂霖
文 杨,蒋小艳,王浩园,范文辉,周茂霖,周 强△
1.重庆市高新区人民医院重症医学科,重庆 400039;2.重庆市第九人民医院神经内科,重庆 400799;3.重庆理工大学药学与生物工程学院,重庆 400054
重症监护室(ICU)患者病情危重,免疫力低下,侵入性操作较多,加之频繁使用抗菌药物,容易出现肺部感染,是医院感染的高危人群[1-2]。肠道菌群对抑制宿主感染、促进炎症反应和维持宿主免疫反应与菌群活性间的平衡至关重要,还可帮助宿主抵抗致病菌,如诱导宿主的免疫交叉反应以产生针对病原体的保护性免疫[3]。研究指出,危重症患者的肠道菌群多样性显著降低,肠道微生态紊乱似乎与不良预后相关[4-5]。另有研究提出了“肺肠轴”的概念,肠道菌群通过微生物分泌物和免疫调控因子对肺部健康产生影响,肠道中的消化副产物可能影响免疫系统从而调控肺部的炎症,而肠道菌群和肺部菌群可能通过淋巴中的液体互相交换[6]。因此,维持肠道菌群稳定对提高ICU肺部感染患者的治疗效果十分重要。本研究使用16S rRNA测序技术分析危重症患者粪便微生物菌群的结构特征,并将肺部感染与非肺部感染的危重患者和健康人群的检测分析结果进行比较,进而寻找危重症患者肺部感染的肠道微生态危险因素,有助于制订新的预防和治疗策略。
1 资料与方法
1.1一般资料 选取2021年3-9月重庆市高新区人民医院ICU收治的50~90岁的危重症患者和体检中心的50~90岁健康人群作为研究对象,包括78例ICU肺部感染患者(Y-ICU组),22例ICU非肺部感染患者(N-ICU组)和40例健康人群(HC组)。ICU肺部感染患者中,未服用抗菌药物的患者48例,服用抗菌药物的患者30例。ICU非肺部感染患者中,未服用抗菌药物的患者15例,服用抗菌药物的患者7例。挑选ICU中未服用抗菌药物的患者作为N-antibiotic组(63例),服用抗菌药物的患者作为Y-antibiotic组(37例)。
Y-ICU组中,男42例,女36例;年龄(75.95±11.46)岁;体质量指数(BMI)(21.37±4.39)kg/m2;急性生理与慢性健康评分系统Ⅱ评分(APACHⅡ评分)(21.03±9.17)分;住院时间的中位数为34.50 d。N-ICU组中,男11例,女11例;年龄(74.77±8.85)岁;BMI(20.33±2.88)kg/m2;APACHⅡ评分(19.14±7.84)分;住院时间的中位数为11.50 d。HC组中,男20例,女20例;年龄(72.03±9.61)岁;BMI(22.11±3.00)kg/m2。Y-ICU组与N-ICU组的年龄、BMI、APACHⅡ评分和住院时间差异均无统计学意义(P>0.05),具有可比性;Y-ICU组、N-ICU组、HC组年龄结构和BMI差异均无统计学意义(P>0.05),具有可比性。
1.2纳入、排除标准 纳入标准:入住ICU的危重症患者。排除标准:年龄<50岁者;存在消化道疾病或行胃肠道手术治疗者;采样前1个月内服用过益生菌或抗微生物制剂者及合并自身免疫性疾病、血液系统疾病者。本研究符合医学伦理学标准,获得重庆高新区人民医院伦理委员会批准,入组前均征得受试者或直系家属知情同意,并签署知情同意书。
1.3方法
1.3.1样本采集 用5 mL粪便采集管(上海朗赋实业有限公司)收集ICU患者入院1 d内的新鲜粪便样本或健康人群的新鲜粪便样本置于干冰上保存,于2 h内转移到-80 ℃冰箱保存,直至提取粪便中微生物基因组DNA。
1.3.2粪便基因组DNA提取与16S rRNA测序 本研究采用FastDNA®Spin Kit for Soil试剂盒(美国MP Biomedicals公司)提取研究对象的粪便基因组DNA。利用1%琼脂糖凝胶电泳(5 V/cm,20 min)检测基因组DNA完整性,用NanoDrop2000检测DNA纯度和浓度。然后利用引物338F(5′-ACTCCTACGGGAGGCAGCA-3′)和806R(5′-GGACTACHVGGGTWTCTAAT-3′)对基因组的16S rDNA的V3~V4区进行第1轮扩增,扩增体系(共20 μL)包含10 ng的DNA模板,0.8 μL的正向引物338-F(5 μmol/L),0.8 μL的反向引物806R(5 μmol/L),4 μL 5×Fast Pfu Buffer,2 μL 2.5 mmol/L dNTPs ,0.4 μL Fast Pfu Polymerase,0.2 μL BSA,并补ddH2O至20 μL。第1轮扩增程序为:95 ℃预变性3 min;95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸45 s,循环27次;72 ℃再延伸10 min,4 ℃冷却保存。第1轮扩增产物选择3 μL 2%的凝胶电泳(120 V,20 min)用于检验目的片段是否成功扩增。随后,用磁珠法纯化第1轮扩增产物。纯化的扩增产物由重庆浦洛通基因医学研究院上机测序。扩增产物加上特异性barcode用于识别样本,16S rRNA V3~V4区扩增产物采用双端测序策略(PE300)。测序反应在Illumina MiSeq测序平台上进行,要求每个样本产生的测序数据不低于50 000条序列片段。
1.3.3生物信息学分析 将Miseq测序下机数据根据特异性barcode序列进行样本拆分,获得每个样本双端测序数据即Paired-end(PE) 序列片段,根据PE序列片段之间的overlap采用Flash软件对数据进行拼接(拼接后的序列称为标签),每个样本不得低于50 000条标签。对标签序列进行质量控制,去除N含量超过0.1%的标签,去除质量得分<20分的低质量标签。基于97%的序列相似性用Usearch将序列归类操作成运算分类单元(OTU),基于SLILVA 16S rRNA基因数据库,用RDP(http://rdp.come.mus.edu/)进行序列的微生物分类(置信度70%),使用统计学分析方法对每一样本进行不同分类级别的分类。根据统计学分析结果,得到每个样本的微生物群落多样性和丰度,并通过箱线图等形式呈现出来。用Mothur分析样本的alpha多样性,其中包含Shannon指数、Chao1指数。利用Bray-Curtis相异指数和两项非参数检验[置换多元方差分析(Adonis分析)和多响应置换过程分析(MRPP分析)]对数据进行相异检验;利用主成分分析(PCA)比较不同样本的菌群结构,用聚类-热图分析不同菌群对治疗策略的响应。采用LEfSe分析(线性判别分析)统计不同组别中有显著作用即差异有统计学意义,微生物类群的LDA得分,展示LDA得分大于设定阈值(LDA得分>2分)的物种。然后,利用Tax4Fun软件预测ICU患者肠道菌群群落的功能。最后,用皮尔逊相关系数矩阵图表征ICU患者肠道菌群与临床指标的相关性。

2 结 果
2.1ICU肺部感染患者肠道菌群的物种多样性比较分析 通过对Y-ICU、N-ICU和HC组进行16S rRNA高通量测序,总共获得8 267 315条有效序列,平均有效长度为401 bp。这些序列以97%的序列相似性聚类为2 540个OTU。ICU患者肠道菌群的物种多样性较健康人差异有统计学意义(P<0.05),与HC组比较,Y-ICU组和N-ICU组Chao1指数均下降(P=0.002、P<0.001),见图1A。与HC组比较,Y-ICU组Shannon指数降低(P=0.002),Y-ICU组与HC组有较为不同的肠道菌群丰富度,见图1B。N-ICU与Y-ICU组Chao1指数和Shannon指数比较差异无统计学意义(P>0.05),Y-ICU和N-ICU组具有相似的菌群丰富度和多样性。
笔者采用Adonis分析探讨Y-ICU与N-ICU和HC的菌群结构是否存在差异,结果显示,Y-ICU组和N-ICU组的肠道菌群结构与HC组比较差异均有统计学意义(P=0.001、0.001),但Y-ICU和N-ICU组肠道菌群结构差异无统计学意义(P=0.907),见图1C。
2.2Y-ICU、N-ICU和HC组肠道菌群门和属水平组成差异分析 选择Y-ICU、N-ICU和HC组优势菌群中相对丰度排名前5位的门水平进行分析,结果显示,HC、N-ICU、Y-ICU组的肠道菌群都以厚壁菌门和拟杆菌门为主,HC组厚壁菌门和拟杆菌门平均相对丰度的总和高于N-ICU组和Y-ICU组(P<0.05)。与N-ICU组比较,Y-ICU组厚壁菌门、变形菌门、梭杆菌门、拟杆菌门和放线菌门的相对丰度差异无统计学意义(P>0.05)。见图2A、B。

注:A为相对丰度排名前5位的门水平差异分析的箱线图;B为相对丰度排名前5位的门水平分析;C为相对丰度排名前10位的属水平差异分析的箱线图;D为相对丰度排名前10位的属水平分析;E为ICU患者单一优势菌群丰度≥50%的样本热图;HC、N-ICU、Y-ICU分别为HC组、N-ICU组、Y-ICU组;ns为P≥0.05,*为0.010≤P<0.05;**0.001≤P<0.01;***P<0.001。
选择Y-ICU、N-ICU和HC组优势菌群中相对丰度排名前10位的属水平进行分析,结果显示,3组的肠道菌群以普雷沃菌属和拟杆菌属为主,且HC组属水平多样性差异有统计学意义(P<0.05)。与HC组相比,排名前10位的菌属中,棒状杆菌属、卟啉单胞菌属、厌氧球菌属、芬戈尔德菌属和嗜胨菌属明显富集于N-ICU组和Y-ICU组(P<0.05)。值得注意的是,双歧杆菌作为最常见的肠道益生菌之一,其相对丰度在Y-ICU组中较N-ICU组更低(1.17%vs.2.35%)。见图2C、D。另外,在HC、N-ICU和Y-ICU组中大部分样本的单一优势菌群相对丰度<50%,但是78例ICU肺部感染患者的样本中,有11例样本的单一优势菌群相对丰度≥50%,表明这些样本存在较严重的肠道菌群失调,且其优势菌群主要集中在普雷沃菌属、拟杆菌属、肠球菌和埃希菌等,见图2E。
本研究在Y-ICU和N-ICU组之间总共筛选到10个相对丰度差异有统计学意义的OTU,其中在Y-ICU组富集的有8个OTU,包括疣微菌科、阿克曼菌属、疣微菌纲、疣微菌目、疣微菌门、月形单胞菌属、克劳芽孢杆菌属、消化链球菌科。在N-ICU组中富集的有2个OTU,为短杆菌属和微球菌属。N-ICU组和Y-ICU组差异最明显的肠道微生物是疣微菌科及其代表菌属阿克曼菌。见图3。
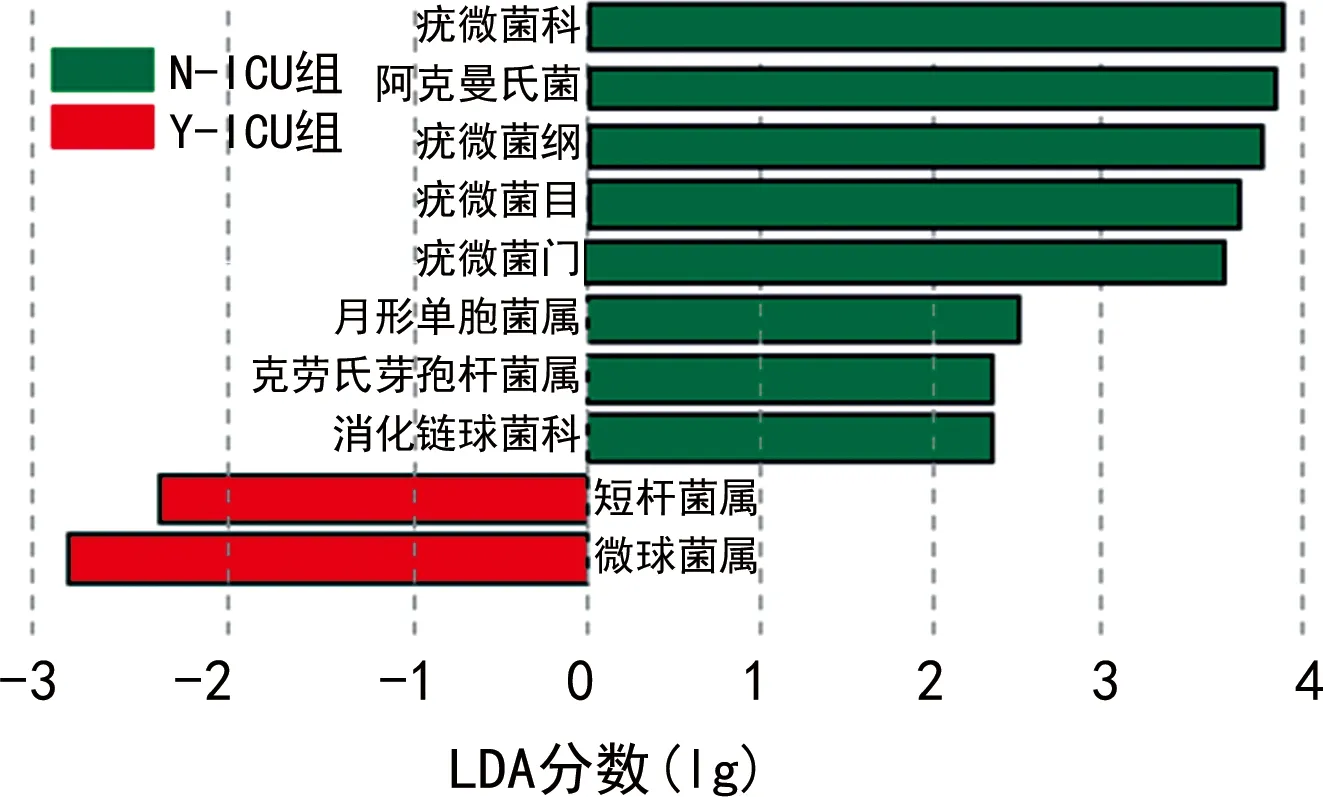
图3 Y-ICU组与N-ICU组基于OTU的LEfSe分析
2.3ICU患者肠道菌群功能预测 共预测获得6 500个KEGG Orthology(KO),可以分类到210条代谢通路中。LEfSe分析显示,ICU患者与健康者比较有4条代谢通路差异有统计学意义(P<0.05,LDA得分>3分),富集的代谢通路主要是基础代谢相关的通路,如氧化磷酸化、RNA降解、ABC转运器、硫继电系统。HC富集的代谢通路主要是与糖类和氨基酸类代谢相关的通路,如戊糖和葡萄糖醛酸转化途径、淀粉和蔗糖代谢、丙氨酸天门冬氨酸和谷氨酸代谢、氨基糖和核苷酸糖新陈代谢等。见图4。此外,在Y-ICU组和N-ICU组中未筛选到差异代谢通路。

图4 基于代谢通路ICU患者和健康者LEfSe分析
2.4ICU肺部感染患者肠道菌群与临床指标的相关性 结果显示,与HC组比较,Y-ICU和N-ICU组厚壁菌门/拟杆菌门的比值增加(P<0.05)。HC组厚壁菌门/拟杆菌门的比值都<10,但是有13例ICU患者厚壁菌门/拟杆菌门的比值>10,其中3例患者该比值更是>100,且厚壁菌门/拟杆菌门比值过大的样本都表现出拟杆菌门丰度极小的倾向。ICU肺部感染者和非肺部感染者的厚壁菌门/拟杆菌门的比值差异无统计学意义(P>0.05)。革兰阳性菌/革兰阴性菌的比值在HC、N-ICU和Y-ICU组中差异无统计学意义(P>0.05)。另外,服用抗菌药物与否的ICU患者尽管肠道菌群的Chao1指数差异有统计学意义(P<0.05),但是Shannon指数在两组间差异无统计学意义(P>0.05)。见图5A、B。
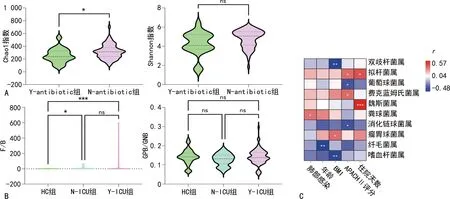
注:A为ICU患者中抗菌药物服用和非抗菌药物服用者肠道菌群alpha多样性;B为HC、N-ICU、Y-ICU组中厚壁菌门/拟杆菌门、革兰阳性菌/阴性菌比值情况;C为患者肠道菌群与临床参数Spearman秩相关系数热图;GPB/GNB分别表示革兰阳性菌/革兰阴性菌的比值;F/B表示厚壁菌门/拟杆菌门的比值;ns为P≥0.05;*0.01≤P<0.05 ;**0.001≤P<0.01;***P<0.001。
笔者评估了年龄结构、BMI、APACHⅡ评分和住院时间等与Y-ICU肠道菌群的关系。结果显示,ICU患者肺部感染仅与粪球菌属有关(P<0.05)。APACHEⅡ评分与拟杆菌属和费克蓝姆菌属呈正相关(P<0.05),与葡萄球菌属和消化链球菌属呈负相关(P<0.05)。住院时间与魏斯菌属呈正相关(P<0.05)。BMI与瘤胃球菌属呈正相关(P<0.05),与双歧杆菌属和嗜血杆菌属呈负相关(P<0.05),见图5C。
3 讨 论
肠道菌群被认为是人体的一个重要“器官”,对营养物质代谢、人体自身发育、免疫、疾病发生和转归均具有重要意义[7]。肠道中定植的厌氧菌、兼性厌氧菌可诱导肠道黏膜引起免疫反应,以抵御病原微生物的入侵,增强宿主的抗病能力,避免肠道受损害[8-10]。危重症患者的微生物组研究方兴未艾,多项研究证实肠道菌群失调在多种危重疾病中发挥着重要作用,如脓毒症、急性肾损伤和多器官功能衰竭等[11-13]。ICU肺部感染的部分患者的肠道菌群可能存在极端生态失调,同时免疫功能低下难以制衡肠道微生物的平衡,从而容易引发感染。危重症患者持续暴露于广泛的内环境改变(如儿茶酚胺产生增加、葡萄糖代谢改变和胃肠功能障碍)以及临床干预措施(如质子泵抑制剂、阿片类药物、营养支持和抗菌药物),这些因素均会影响肠道菌群组成,而紊乱的肠道菌群有助于病原菌的生长[14]。因此,在危重症患者治疗过程中,结合肠道菌群研究和临床数据,综合分析宿主和微生物间的相互作用,以及环境因素的影响,可对其治疗提供积极的建议。通过对驱动微生物群落发育的生态和环境因子、菌群如何影响肺部感染的研究,可以为危重症患者康复过程提供临床指导,并具有重要的诊断价值。
本文通过对ICU患者和健康人群进行16S rRNA高通量测序分析,发现ICU肺部感染患者肠道菌较健康人群Chao1指数显著降低,肠道菌群显著失调,这与以往研究结果一致[11-13]。通过分析健康人群、危重症肺部感染和非肺部感染患者的肠道菌群组成发现,危重症肺部感染患者肠道中双歧杆菌(促进健康的共生微生物)的丰富度略有降低,而变形菌门(病原微生物)的丰富度高于健康人群,这与之前研究ICU患者肠道微生物群落的特征一致[15]。另外,在肺部感染患者中微生物极度失调的患者(单一菌群相对丰度≥50%)的肠道菌群主要富集了普雷沃菌属、拟杆菌属、肠球菌和埃希菌。其中普雷沃菌属是一种潜在的致病菌,可参与局部和全身感染,常引起口腔、肌肉骨骼、头颈部、皮肤、下呼吸道和肺部等感染[16-17]。而肠球菌和埃希菌是ICU新生儿的主要早期肠道菌群[18],埃希菌感染也可通过调节JAK/STAT信号,诱导炎症因子——抗菌药物α(IFN-α)和抗菌药物β(IFN-β)的产生,最终造成肺部炎症或者损伤[19]。而主要富集在ICU患者中的芬戈尔德菌属、嗜胨菌属和卟啉单胞菌属是糖尿病足骨髓炎感染的主要病原菌[20]。这些证据表明了ICU肺部感染患者肠道菌群中与感染相关的病原菌的丰度增加,从一定程度上解释了入住ICU的患者有更大的感染风险。而ICU肺部感染者中肠道菌群极度失调者造成的肺部炎症似乎由肠道菌群紊乱引起的自身免疫系统诱导炎症因子所引起。本文的ICU患者肠道微生物群落的功能预测表明ICU患者肠道微生物糖类和氨基酸类代谢潜力低于健康人群,而糖代谢和氨基酸代谢能力的下降,会使危重症患者的内环境改变,从而影响肠道菌群的组成。因此危重患者需要补充肠内营养或肠外营养来平衡失调的肠道微生态,以提高免疫力。
本研究在探索常见肠道菌群参数与ICU肺部感染结局的关系时,结果显示革兰阳性菌/革兰阴性菌的比值在HC、N-ICU和Y-ICU组中并无明显差异,表明革兰阳性菌/革兰阴性菌的比值不足以作为ICU肺部感染患者肠道菌群的特征性指标。厚壁菌门/拟杆菌门这一指标虽然在健康人和ICU患者中差异有统计学意义,但是ICU肺部感染者和非肺部感染者的厚壁菌门/拟杆菌门的比值并无明显差异,因此认为厚壁菌门/拟杆菌门作为危重症患者中肺部感染者的标志性指标欠缺说服力。另外本研究结合年龄结构、BMI、APACHⅡ评分和住院时间等评估ICU肠道菌群的关系,笔者发现未来拟杆菌属、费克蓝姆菌属、葡萄球菌属、消化链球菌属和魏斯菌属这些指标对提示预后和治疗进展具有参考价值。但是由于本研究采用的临床样本数量有限,是否能把上述菌属作为临床危重症的评价指标还需进一步扩大临床样本数量进行验证。
综上所述,本研究发现与健康人群相比,ICU肺部感染患者肠道菌群多样性显著降低,微生态失调,病原菌增加,促进健康的共生微生物减少。而ICU肺部感染患者肠道菌群的失调也许与糖代谢和氨基酸代谢下降有关。另外,基于肺肠轴理论和本研究的结果,ICU肺部感染患者可能会通过特殊菌属(普雷沃菌、肠杆菌和埃希菌等)的感染引发自身免疫系统诱导炎症因子产生并最终造成肺部感染,提示肠道菌群失调与ICU肺部感染有密切关系,也为ICU肺部感染患者的微生态靶向治疗提供了一定的依据。
