基于毒性表型和基因型的主要冬繁区小麦条锈菌群体遗传分析
2023-08-15高新培赵鋆刘博凡郭一康振生詹刚明
高新培,赵鋆,刘博凡,郭一,康振生,詹刚明
基于毒性表型和基因型的主要冬繁区小麦条锈菌群体遗传分析
高新培,赵鋆,刘博凡,郭一,康振生,詹刚明
西北农林科技大学植物保护学院/旱区作物逆境生物学国家重点实验室,陕西杨凌 712100
【目的】明确中国主要冬繁区小麦条锈菌群体毒性结构和遗传多样性,为冬繁区及黄淮海麦区小麦条锈病的防控及小麦抗性基因的合理布局提供参考依据。【方法】从四川盆地、湖北和河南南部等主要冬繁区采集并分离得到148个小麦条锈菌菌株,利用中国鉴别寄主和单基因系鉴别寄主进行毒性表型鉴定;并利用17对KASP-SNP引物对菌株进行标记,完成基因型分析。【结果】基于中国鉴别寄主共鉴定出14个已知小种和63个未知致病类型,其中CYR34(16.2%)、G22-14(12.2%)、CYR32(6.8%)、CYR33(5.4%)为优势小种(致病类型);基于单基因系鉴别寄主鉴定得到113个小种(致病类型),其中race1(7.4%)、race2(3.4%)、race3(3.4%)为优势小种(致病类型)。贵农22类群是中国冬繁区小麦条锈菌群体的最大流行类群,供试条锈菌均不侵染携带和的单基因系品种。单基因系毒性鉴定及分子标记均显示CYR34和G22-14的毒性表型及基因型呈现多样化,表明这两个优势小种内部存在高度分化。基于两套鉴别寄主的毒性数据聚类显示,四川盆地与湖北南部条锈菌群体相似,而湖北西北部与河南南部条锈菌群体相似;基于KASP-SNP分子数据的遗传聚类显示,四川盆地、湖北南部条锈菌群体与湖北西北部、河南南部条锈菌群体基因型存在分化;Structure分析显示四川盆地、湖北南部群体主要有2种遗传背景,湖北西北部、河南南部群体主要有一种遗传背景;群体遗传分化分析显示四川盆地条锈菌群体与河南南部条锈菌群体二者值最大,为0.118,遗传差异最大且遗传分化明显;湖北西北部群体与河南南部群体遗传分化程度最小,值为0.010;基因流分析得到湖北西北部群体与河南南部群体之间的m值为25.236,m>4,二者存在较大的基因流,湖北西北部和河南南部群体与四川盆地群体之间的m值分别为2.923和1.864,均存在较小的基因流;遗传多样性分析结果表明,四川盆地、湖北南部地区条锈菌群体遗传多样性水平均较高,湖北西北部、河南南部条锈菌群体遗传多样性水平较低。上述结论均支持四川盆地、湖北南部群体同湖北西北部、河南南部群体存在遗传分化。【结论】单基因系鉴别寄主能够精准地进行中国小麦条锈菌小种鉴定;中国主要冬繁区的小麦条锈菌群体存在不同来源。
小麦条锈菌;冬繁区;毒性鉴定;KASP-SNP;群体遗传
0 引言
【研究意义】由条形柄锈菌小麦专化型(f. sp.,)所引致的小麦条锈病,是小麦生产上最具毁灭性的病害之一[1-2],严重威胁着中国的小麦安全生产[3-4]。加强冬繁区小麦条锈菌群体的监测、明确小麦产区条锈菌个体毒性特征及群体毒性结构并揭示小麦条锈病的传播流行规律对于防止春季流行区病害的发生、制定合理有效的病害防控策略至关重要。【前人研究进展】前人研究表明,中国小麦条锈病分为三大流行区系[5-6],其中,冬繁区是中国春季小麦条锈病流行的菌源基地,在越夏易变区和春季流行区之间起着桥梁作用。研究报道,2016年秋河南省南部及湖北省北部麦区接受了来自甘肃东南以及陕西关中等地的菌源,使得小麦条锈病在江汉平原扩散[7]。Huang等[8]对2017、2018年中国中西部多个省份的条锈菌菌株进行研究,发现鄂西北与鄂南的条锈菌存在不同的来源。Zhan等[9]通过群体遗传分析,发现2019年湖北地区的小麦条锈菌来源于云南东部和贵州。【本研究切入点】目前,关于冬繁区小麦条锈菌群体结构及其菌源的研究鲜见报道,而冬繁区小麦条锈菌的小种和致病类型构成在相当大程度上代表了黄淮海麦区春季流行的群体构成。因此,及早监测冬繁区小麦条锈菌群体遗传结构对针对性防控春季流行区病害具有关键意义。【拟解决的关键问题】本研究利用中国鉴别寄主和单基因系鉴别寄主两套鉴别寄主,并利用17对竞争性等位基因特异性单核苷酸多态性(kompetitive allele specific PCR-single-nucleotide polymorphism,KASP-SNP)引物,研究中国四川盆地、湖北南部、湖北西北部和河南南部地区的小麦条锈菌群体毒性结构和遗传多样性,明确冬繁区群体的毒性特征及遗传关系,旨在明确冬繁区小麦条锈病的传播流行规律,为冬繁区及黄淮海麦区小麦条锈病的防控及小麦抗性基因的合理布局提供参考依据。
1 材料与方法
1.1 材料
小麦条锈菌标样:2020年1月在四川盆地(SCB)、湖北南部(S-HB)、湖北西北部(NW-HB)和河南南部(S-HN)采集小麦条锈菌标样,共繁殖存活148份。其中,四川盆地81份,湖北南部25份,湖北西北部26份,河南南部16份(表1)。

表1 2020年冬繁区标样采集信息表
供试小麦品种:19个中国鉴别寄主依次为Trigo Eureka、Fulhard、保春128、南大2419、维尔、阿勃、早洋、阿夫、丹麦1号、尤皮Ⅱ号、丰产3号、洛夫林13、抗引655、水源11、中四、洛夫林10、Hybrid 46、和贵农22,对照品种铭贤169(高度感病),均由西北农林科技大学旱区作物逆境生物学国家重点实验室提供(电子附表1)。小麦条锈菌18个单基因系及5个辅助鉴别寄主材料(电子附表2)由美国华盛顿州立大学陈贤明教授惠赠。
1.2 标样分离与菌系扩繁
将铭贤169种植于10 cm×10 cm×10 cm(长×宽×高)的花盆中,每盆播种12—15粒种子,待第一叶完全展开时接种。用流水冲去标样叶片上的泥土和杂质,甩干,将叶片正面朝上,放入铺有湿润滤纸的托盘中,10 ℃黑暗保湿8—10 h。用无菌大头针挑取标样上的单个夏孢子堆均匀涂抹于脱蜡后的幼苗叶片正面,保证每个幼苗叶片只接种1个夏孢子堆,每盆幼苗只接种1个标样。接种后于10 ℃黑暗保湿24 h,而后再转移到温室培养,培养条件为17 ℃ 17 h光照/13 ℃ 7 h黑暗。待接种叶片出现明显褪绿斑时,每盆只留1株发病较好的幼苗并加上隔离罩,防止不同菌系间的交叉污染。接种后13—15 d,用干净试管分别收集病叶上的夏孢子,置于4 ℃干燥器内保存备用或于-80 ℃长期留存备用。
1.3 菌系的毒性表型鉴定与毒性分析
将两套鉴别寄主的小麦品种编号,按顺序依次种植19个中国鉴别寄主、18个单基因系和5个辅助鉴别寄主,以及对照品种铭贤169,每个花盆的4个角落种植4个品种,并插上标签牌以作标记。在鉴别寄主幼苗一叶完全展开后,均匀喷洒展布剂0.1%吐温20溶液,将足量夏孢子分别与干燥的滑石粉按1﹕20的比例混合,均匀接种至两套鉴别寄主及铭贤169(对照)的叶面上,喷洒少量水雾,置于黑暗湿润的保湿间中10—12 ℃保湿24 h,再转入温室培育17—21 d,按照9级分级标准[10],记录各菌种在鉴别寄主上的反应型,0—6级为无毒性,7—9级为毒性。无毒性数据转化为0,毒性数据转化为1,用Excel统计条锈菌群体对各鉴别寄主的毒性频率。
1.4 菌系夏孢子DNA的提取及KSAP-SNP标记
参照Aljanabi等[11]的CTAB法提取条锈菌夏孢子DNA,并稍加改进。用分光光度计检测DNA的浓度和纯度,并将其稀释至标准液50 ng·μL-1备用。
实验室前期完成了特异性KASP-SNP引物的设计和筛选工作[12],通过进一步筛选得到17对对条锈菌群体具有多态性的KASP-SNP引物(表2),用于本研究。
取DNA样品稀释液2 μL于384孔板底部并烘干;配置KASP反应体系(5 µL):2×KASP Mastermix 2.5 μL、primer mix(Forward primer 12 mmol·L-1和Reverse primer 30 mmol·L-1)0.056 μL和ddH2O 2.444 µL。

表2 17对具有多态性的KASP-SNP引物
采用Touchdown方法进行PCR扩增:94 ℃ 15 min;94 ℃ 20 s,65 ℃ 60 s,每个循环退火温度降0.8 ℃,10个循环;94 ℃ 20 s,57 ℃ 60 s,32个循环。PCR反应结束后,用酶标仪读取384孔板中的荧光数据(数据读取在40 ℃以下进行)。对荧光信号较低的数据,可适量增加循环后再次读取,但总循环数不能超过48个,除去缺失数据结果共得到138株菌株数据结果。(具体操作步骤参考LGC官网)。
1.5 数据分析
用毒性分析软件VAT[13]统计毒性多样性指数、毒性表型特征及毒性频率(病原菌群体对特定抗性基因的侵染频次)。用Structure2.3.4软件分析KASP-SNP分子标记结果[14],设置运行参数K(2—5),10次重复,利用Structure Harvester确定最佳K值,利用Clusters软件重复抽样分析[15],利用distruct软件绘制结果图在Adobe Illustrator CS5展现[16];利用POPGENE计算基于分子数据群体间的遗传距离、基因流m、遗传分化指数、观察等位基因数a、有效等位基因数e、香农信息指数、Nei’s遗传多样性指数s、多态性位点数、多态性位点百分比;使用Powermarker V3.25计算遗传距离,并用MGEA5.2生成菌株个体聚类图;根据得到的毒性数据与分子数据,利用R软件中的2.5.0软件包的层次聚类分析基于Nei’s遗传距离采用UPGMA生成聚类树,自展值(Bootstrap value)通过1 000次的重复计算所得;利用NTSYS-pc软件中的MXCOMP矩阵比较程序对分子基因型与毒性表型进行Mantel相关检验分析[17]。使用R语言程序包2.5.0进行连锁不平衡分析[18]。
2 结果
2.1 供试菌系的毒性鉴定结果
2.1.1 冬繁区小麦条锈菌生理小种分析 根据中国鉴别寄主鉴定结果(表3),供试冬繁区148份小麦条锈菌分属于4个不同类群:即贵农22致病类群(频率为40.0%)、杂种46致病类群(频率为29.7%)、水源11致病类群(频率为18.9%)和洛夫林13致病类群(频率为8.8%)。4个类群共计占所鉴定菌株总数的97.0%以上;另发现古老生理小种CYR24各1株,出现在四川盆地和湖北南部。
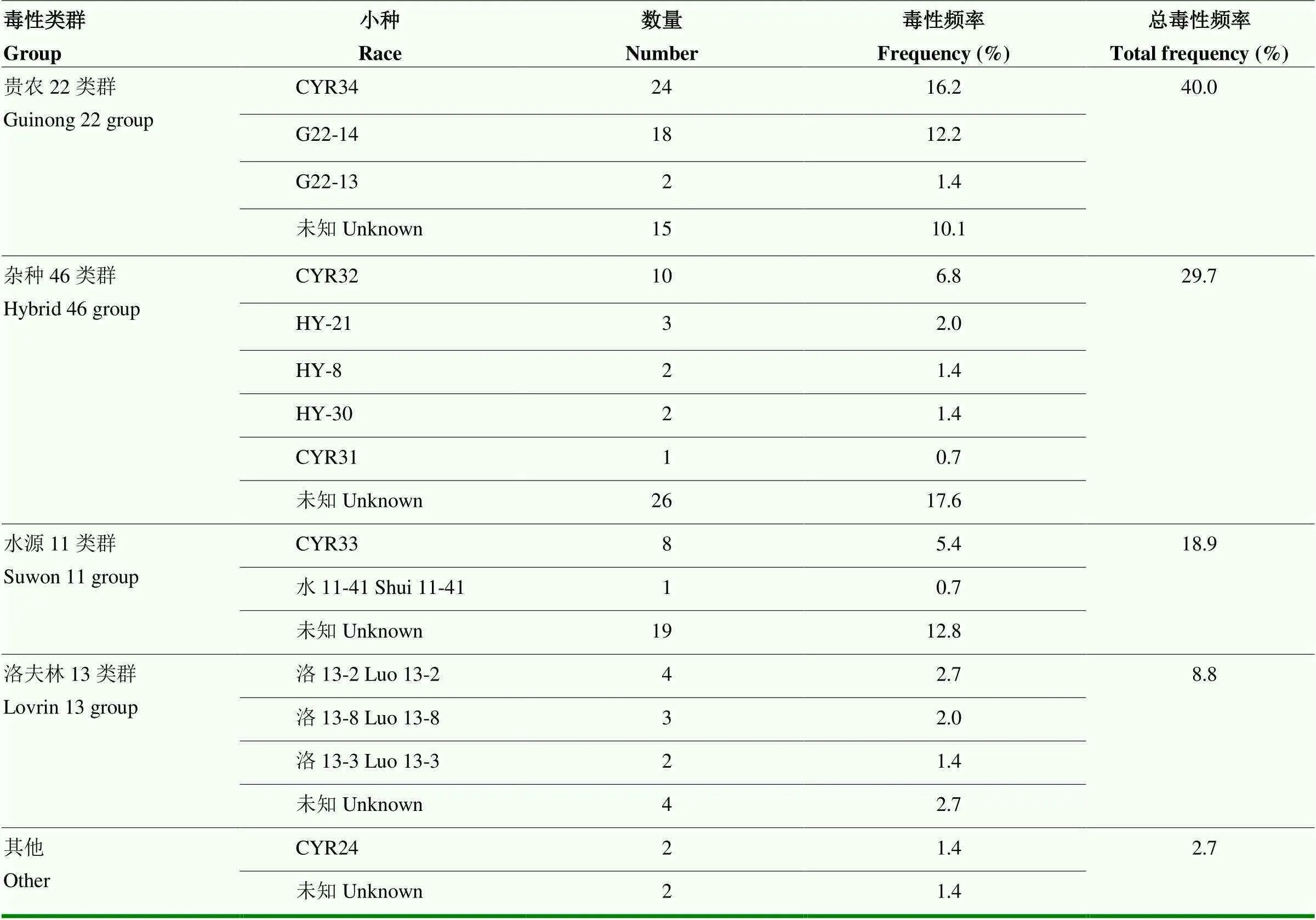
表3 生理小种鉴定结果
共鉴定出14个已知条锈菌小种,其中,CYR34有24株菌株,频率为16.2%,位居首位;G22-14有18株菌株,频率为12.2%,居于第二位;CYR32有10株菌株,频率为6.8%,居第三位;CYR33有8株菌株,频率为5.4%,居第四位。这4个生理小种出现频率总和占比40.6%,其余生理小种各自所占频率均小于3.0%。
从分布地区来看(表4),在四川盆地排名前三的小种是:CYR34(9.9%)、G22-14(9.9%)和CYR32(4.9%);在湖北南部排名前三的小种是:CYR34(28.1%)、G22-14(12.5%)和CYR32(12.5%);在湖北西北部排名前三的小种是:CYR34(20.0%)、G22-14(15.6%)和CYR33(11.1%);在河南南部排名前三的小种是:CYR34(18.8%)、G22-14(18.8%)和G22-13(12.5%)。

表4 各地区出现频率前三的小种类型
从单基因系鉴别寄主鉴定结果来看(电子附表3),148个菌分为113个小种。其中,频率最高的是race1,达7.4%,共计11株菌株;race2和race3出现频率均达3.4%,各有5株菌株;race4—race6出现频率达2.0%,各有3株菌株;race7—race17出现频率均达1.4%,各有2株菌株;剩余其他96种小种频率都为0.7%,各有1株菌株。
从分布地区来看,四川盆地群体出现频率最高的小种为race1和race2(6.2%),湖北南部群体的小种出现频率均为4.0%,湖北西北部群体出现频率最高的小种为race1(23.1%),河南南部群体出现频率最高的小种为race1(18.8%)。race1在湖北西北部及河南南部地区出现频率均为第一,全部表型鉴定结果见电子附表3。以上结果表明,race1出现频率逼近10.0%,存在流行趋势。
综上,贵农22类群是中国冬繁区小麦条锈菌群体的最大流行类群,CYR34出现频率位居所有小种首位;G22-14出现频率超过10.0%已经造成流行,通过单基因系鉴定出的优势小种race1有发展流行风险。
2.1.2 冬繁区小麦条锈菌毒性频率分析 冬繁区148份小麦条锈菌菌株对中国鉴别寄主的毒性频率范围为0—94.6%(图1)。冬繁区的条锈菌均不能侵染中四和。毒性频率在90%—100%的有C2(93.2%)、C6(93.2%)、C8(94.6%)、C11(91.9%),这4种鉴别寄主几乎丧失鉴别能力;毒性频率在70%—90%的有C1(79.1%)、C3(89.2%)、C4(87.8%)、C5(77.7%)、C7(89.2%)、C9(71.6%)、C10(78.4%)、C12(75.0%)、C14(83.8%)和C16(80.4%),这些鉴别寄主毒性差异区分能力不高;其余毒性频率相对较低的是C13(58.8%)、C17(52.0%)和C19(39.9%)。冬繁区小麦条锈菌群体对19个中国鉴别寄主中的大多数具有较高毒性频率。
148份小麦条锈菌菌株对单基因系的毒性频率(图2)范围为0—96.6%。其中,条锈菌群体对跟的毒性频率是0.0;对(96.6%)、(91.9%)、(92.6%)、(92.6%)、(94.6%)、(90.5%)、(95.9%)的毒性频率都在90%以上,对的毒性频率为最高,达到了96.2%;对(89.2%)、(89.9%)、(82.4%)、(85.8%)的毒性频率都在80%以上,对(78.4%)、(79.1%)的毒性频率达到了70%以上;对(63.5%)、(68.9%)、(56.8%)、(61.5%)的毒性频率高于50%而低于70%;对(30.4%)、(31.1%)、(24.3%)和(8.1%)的毒性频率均在40%以下。就18个单基因系及5个辅助鉴别寄主鉴定结果来看,鉴别寄主被侵染频率达到70%以上的有13个,占比56.5%。说明半数以上的单基因系鉴别寄主已经丧失抗性;携带、、和的鉴别寄主具有较好抗锈性,携带与的鉴别寄主表现出高抗性水平。

SCB:四川盆地;S-HB:湖北南部;NW-HB:湖北西北部;S-HN:河南南部。下同
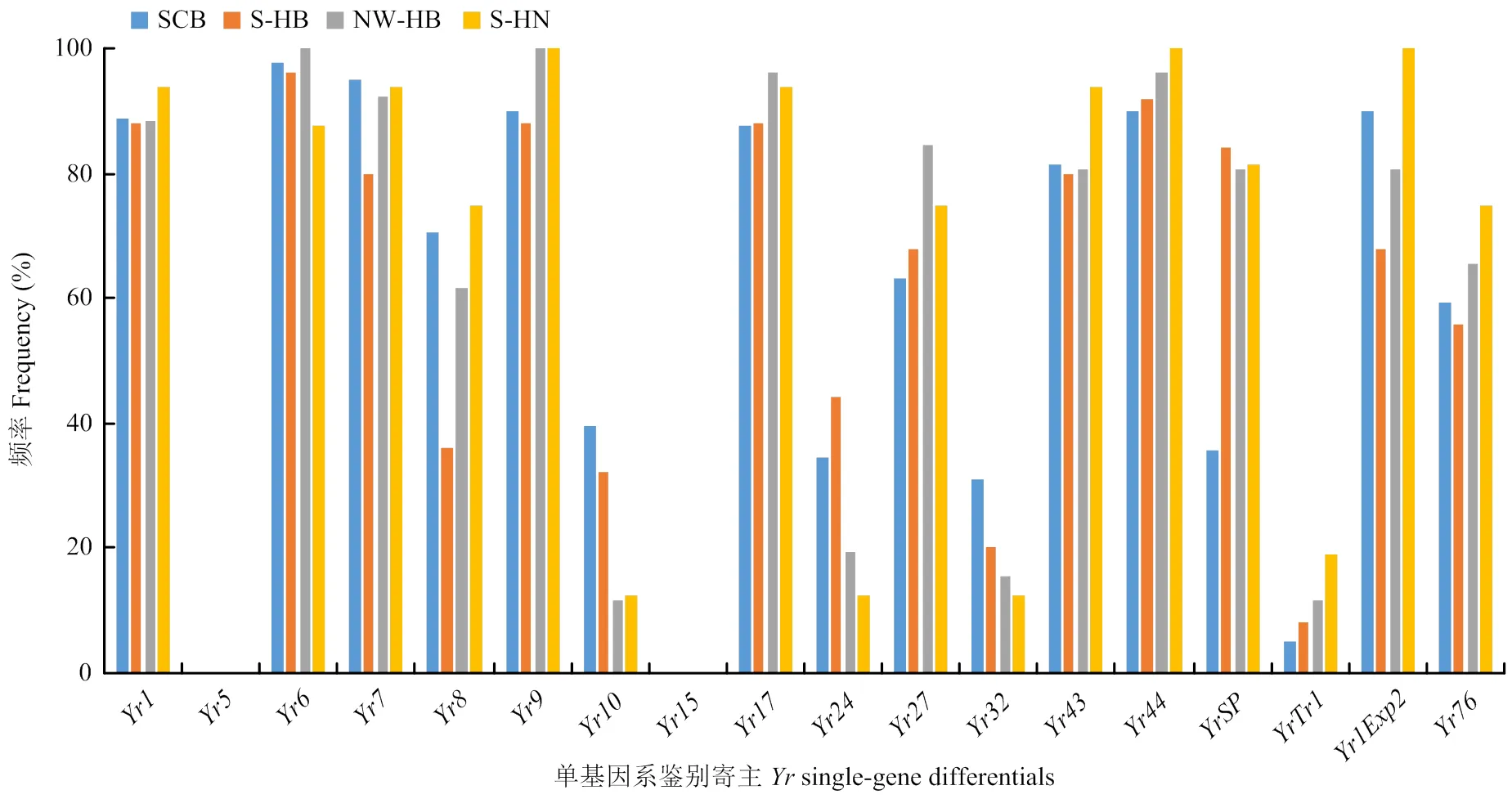
图2 条锈菌群体对单基因系的毒性频率
2.2 冬繁区不同地区条锈菌群体聚类分析
基于中国鉴别寄主毒性数据,利用遗传相似系数构建表型聚类图(图3-A),结果显示,冬繁区群体遗传相似系数范围为0.961—0.980,当遗传相似系数为0.961时,群体可分为2大组。A组:第一亚组为四川盆地、湖北南部群体,第二亚组的群体是湖北西北部群体。B组为河南南部群体。从图3可以看出,四川盆地与湖北南部群体结构最为相近,它们之间可能存在较为密切的菌源交流,但与河南南部相对差异较大,且地理位置也较远,地理距离可能是影响两地区间菌源交流的重要原因。
基于单基因系鉴别寄主毒性数据聚类图(图3-B),结果显示,冬繁区遗传相似系数范围为0.958— 0.971,当遗传相似系数为0.958时,菌株可分为2大组。A组是四川盆地、湖北南部群体。B组是湖北西北部、河南南部群体。从图可以看出湖北西北部跟河南南部群体最为相近,同时四川盆地与湖北南部群体聚为一类。对比两幅聚类图,四川盆地跟湖北南部群体均被聚为一类,具有较高相似度。以上结果表明,四川盆地和湖北南部条锈菌群体毒性结构相似,可能有相同的来源。
通过KASP-SNP分子标记,除去缺失数据结果共计得到138株菌株数据。通过Structure Harvester得到最佳K值为2,138个菌株分属于2个类群(图4)。湖北西北部(NW-HB)与河南南部(S-HN)遗传结构相对简单;四川盆地(SCB)与湖北南部(S-HB)、湖北西北部(NW-HB)与河南南部(S-HN)分别有相似的遗传背景,但存在差异。结果表明,四川盆地与湖北南部群体同湖北西北部与河南南部群体存在遗传分化,前者主要有2种遗传背景,后者主要有1种遗传背景。
分子聚类分析结果显示(图3-C),在0.913处被分为2组,A组是四川盆地与湖北南部条锈菌群体,B组是湖北西北部和河南南部条锈菌群体。A组与B组基因型存在分化,这与Structure所得结论一致。

A:基于中国鉴别寄主的毒性聚类分析;B:基于单基因系鉴别寄主的毒性聚类分析;C:基于SNP标记的聚类分析

图4 不同地区条锈菌群体结构的Structure分析
2.3 CYR34和G22-14的分化比较
共得到24株CYR34菌株,其在单基因系上出现表型分化(图5-A),分化出20个致病类型,最高出现频率为12.5%。当相似性系数为0.727时,菌株被分为2个毒性类群;当相似性系数为0.864时,菌株被聚为5个类群。表明CYR34类群毒性分化复杂。
共得到18株G22-14菌株,其在单基因系上出现表型分化(图5-B),分化出15个致病类型,最高频率为11.1%,有3种表型。当相似性系数为0.707时,菌株被分为2个毒性类群;当相似性系数为0.854时,菌株被聚为5个类群。表明G22-14毒性分化复杂。
在138株菌株的分子数据中,共有22株CYR34和17株G22-14菌株,分别进行基因型个体聚类分析(图6),结果表明,22株CYR34共计15个基因型,17株G22-14共计10个基因型,二者内部均存在遗传分化。
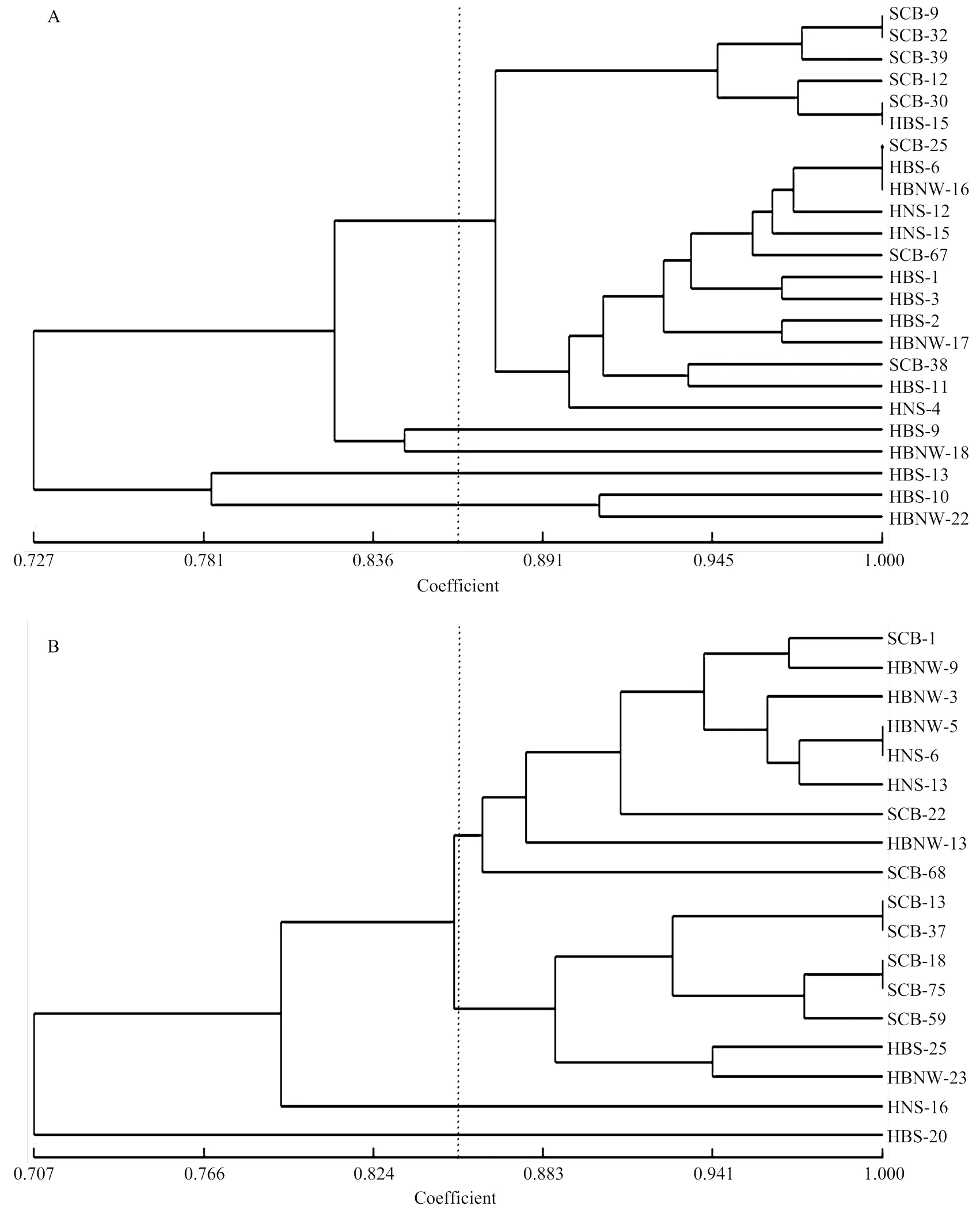
A:CYR34基于单基因系鉴别寄主的毒性聚类分析;B:贵22-14基于单基因系鉴别寄主的毒性聚类分析
2.4 群体遗传分化及基因流分析
遗传分化分析结果表明(表5),四川盆地群体平均值0.072,湖北南部群体平均值0.030,湖北西北部群体平均值0.050,河南南部平均值0.055。相比较而言,四川盆地条锈菌群体与河南南部条锈菌群体二者值最大,为0.118,遗传差异最大且遗传分化明显。湖北西北部群体与河南南部群体遗传分化程度最小,值为0.010。
从m值来看,湖北西北部群体与河南南部群体之间的m值为25.236,m>4,表明二者存在较大的基因流[19],湖北北部和河南南部群体与四川盆地群体之间的m值分别为2.923和1.864,二者存在较小的基因流。四川盆地与湖北南部群体同湖北西北部与河南南部群体存在遗传分化,以上结果与Structure的分析结果指向一致。

图6 CYR34和G22-14的个体聚类分析
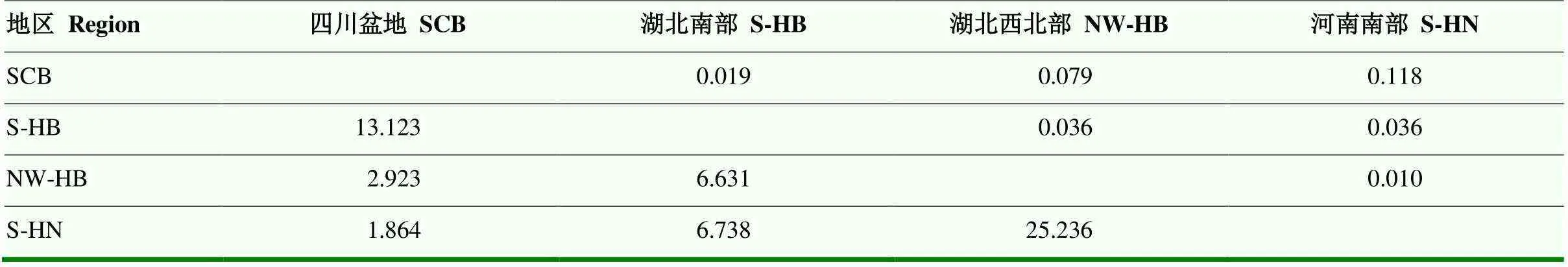
表5 四地区条锈菌群体的遗传分化值Fst(上对角线)和Nm值(下对角线)
2.5 遗传多样性指数分析
遗传多样性统计分析结果显示(表6),在各地区群体之间a范围为1.647—2.000,e为1.523— 1.724,为0.396—0.591,s为0.277—0.406,四川盆地、湖北南部地区、湖北西北部、河南南部依次为17、17、11和11,分别为100%、100%、64.7%和64.7%,表明四川盆地及湖北南部地区条锈菌群体遗传多样性水平均较高,湖北西北部与河南南部条锈菌群体遗传多样性水平较低。其中,四川盆地地区条锈菌群体多态性最高,为0.591,s为0.406,湖北西北部条锈菌群体多态性最低,为0.396,s为0.277。
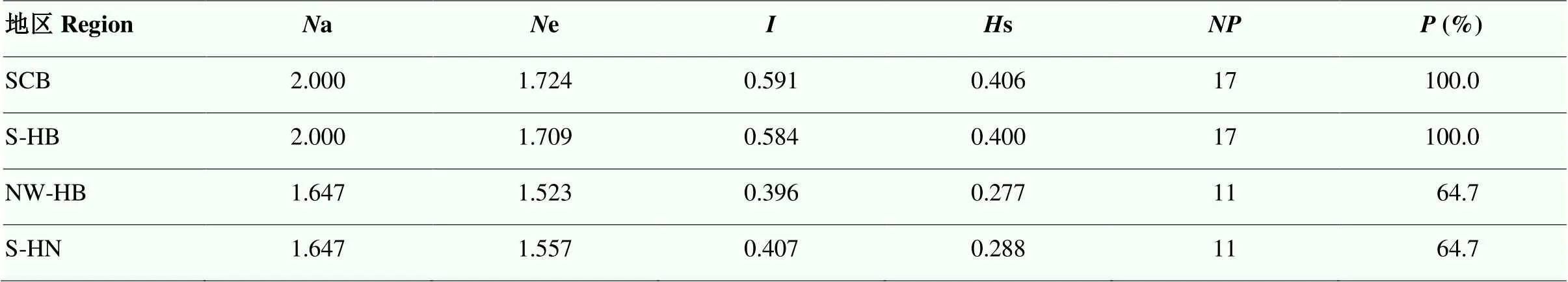
表6 遗传信息指数
a:观察等位基因数;e:有效等位基因数;:香农信息指数;s:Nei’s遗传多样性指数;:多态性位点数;多态性位点百分比
a: observed number of alleles;e: effective number of alleles;: Shannon’s information index;s: Nei's genetic diversity;: number of polymorphic loci;: percentage of polymorphic loci
2.6 冬繁区条锈菌群体繁殖模式分析
小麦条锈菌群体中存在无性生殖跟有性生殖2种繁殖方式,用所得SNP分子标记数据进行连锁不平衡分析,结果显示,所有群体的标准关联指数值显著(<0.01)(图7),这表示连锁平衡的假设不成立,说明这些群体中未发生随机交配或有性生殖。推测这些地区条锈菌群体繁殖模式为无性繁殖。

图7 连锁不平衡分析
2.7 基因型数据与毒性数据相关性分析
利用NTSYS-pc的MXCOMP矩阵比较程序对138株菌株的基因型数据与毒性表型数据进行Mantel相关性检验分析。结果显示,KASP-SNP所得基因型数据与中国鉴别寄主毒性数据相关性极低(1=-0.001,=0.489),与单基因系及辅助鉴别寄主毒性数据相关性极低(2=0.061,=0.955),1和2均接近于0,说明基因型多态性与毒性多态性这两者间的相关性很低,表明基于分子标记的基因型与基于毒性标记的表型2种标记体系之间相互独立。
3 讨论
3.1 冬繁区小麦条锈菌群体结构分析
本研究从四川盆地、湖北和河南南部等主要冬繁区采集并分离得到148个小麦条锈菌菌株,利用中国鉴别寄主和单基因系鉴别寄主对这些分离株进行毒性表型鉴定。同时,利用17对KASP-SNP引物对供试菌株进行分子标记,明确了冬繁区小麦条锈菌群体的流行致病类型、生理小种结构及毒性频率与分布,分析了冬繁区各地区间条锈菌群体遗传结构的差异、基因交流程度及遗传多样性水平。通过毒性表型鉴定与基因型分析,发现冬繁区小麦条锈菌群体存在分化,表明它们可能有不同的来源。
毒性表型鉴定与分子标记聚类分析结果表明,四川盆地与湖北南部条锈菌群体遗传结构高度相似,湖北西北部与河南南部条锈菌群体遗传结构高度相似;Structure分析结果表明,四川盆地及湖北南部地区有两种遗传背景,而湖北西北部与河南南部有近乎单一的遗传背景;从基因流和遗传多样性分析来看,湖北西北部与河南南部群体间存在较大的基因流[19],两群体交流频繁,而二者与四川盆地群体存在较小基因流。这可能是由于地理距离相对较远影响了群体交流,但也说明四川盆地、湖北南部群体与湖北西北部、河南南部群体存在遗传分化[20],从而推测产生这种分化的原因是传播至四川盆地与鄂西北、豫南地区群体的菌源不同,表明冬繁区除接受西北越夏区的菌源以外,还可能接受其他地区的菌源。
甘肃等西北越夏区被认为是小麦条锈菌最重要的菌源地,因此西北越夏区的小麦条锈病历来备受关注与防控。但近年研究表明,西南越夏区不容忽视。Wang等[21]2010年对四川、云南和贵州的越冬和越冬过程进行了研究,指出三地区的菌源极有可能对中国北部、西北和西南部麦区小麦条锈病造成影响。Huang等[8]对中国中西部多个省份2017、2018年的条锈菌进行研究,揭示了条锈菌从云贵地区向湖北地区传播的新路径,鄂西北的条锈菌来自于西北的甘肃地区,而鄂南的条锈菌来自于云贵高原。Zhan等[9]通过群体遗传分析,发现2019年湖北地区的条锈菌来源于贵州和云南东部。本研究表明2020年冬繁区菌源可能不仅来自于西北越夏区,而且可能来自西南越夏区。云贵高海拔地区由于其凉爽的气候和耕作方式,小麦条锈菌可在自生麦苗上越夏,并随气流向其他地方传播。通过对云贵高原八年的气象轨迹数据进行分析,发现气流最远可到达长江中下游地区[22],这表明云贵地区的条锈菌可传播到其他小麦种植区。过去研究认为西南越夏区条锈菌群体只在当地小麦条锈病周年循环中发挥作用,但对全国小麦条锈病的发生与流行有何作用尚不清楚[4],因此后续研究可依据条锈病在田间发病时间的先后采样,用冬繁区条锈菌群体与相关越夏区菌源进行比较分析,进一步探明冬繁区小麦条锈菌来源,为越夏区、冬繁区乃至全国小麦条锈病的精准防控提供依据。
3.2 冬繁区小麦条锈菌流行类群分析
中国鉴别寄主鉴定结果显示贵农22类群是冬繁区的最大流行类群,其大范围流行的原因主要是由于携带(被认为与是相同的)小麦品种的广泛种植[23],使得V26(能克服携带品种抗性的小麦条锈菌)迅速成为优势类群,导致其近年出现频率居高不下且已接近50%。值得注意的是,G22-14的出现频率已经达到了12.2%,比黄瑾等[24]报道的高一倍。结合本研究所得毒性频率结果,发现G22-14与CYR34对单基因系鉴别寄主有相近的毒性频率。G22-14出现频率的快速增长不仅给小麦生产安全带来极大的威胁,而且表明小麦条锈菌群体存在连续不断的毒性变异与毒性分化。中国鉴别寄主毒性频率分布显示:冬繁区小麦条锈菌只对中四和毒性频率为0,对贵农22毒性频率逼近50%,存在抗性丧失风险,这表明中国需要改变以贵农系小麦品种为主的种植策略;单基因系鉴别寄主毒性频率分布显示:条锈菌群体对(30.4%)、(31.1%)、(24.3%)、(8.1%)的毒性频率均在40%以下,对和的毒性频率为0,表明以上基因可以作为有效抗源应用于抗病育种工作。Bai等[25]在2018年对CYR32、CYR33、CYR34和G22-14的寄生适合度测试后,指出小麦育种应减少对和的使用,再结合目前贵农22类群的流行现状,和应被谨慎使用。Huang等[26]测试了2000年到2016年在黄淮海麦区广泛种植的66个商业小麦品种的抗锈基因,发现66个小麦品种均不含有和。结合本试验结果来看,若小麦条锈病在冬繁区发生大流行,积累了足够的菌源量,在春季流行期可轻松攻破黄淮海麦区的小麦抗性防线,引起条锈病的大爆发,严重威胁小麦生产安全。因此,冬繁区抗性基因的精准布局十分重要。
3.3 冬繁区小麦条锈菌毒性分化评价
CYR34与G22-14的毒性聚类图也证实了生理小种内部存在明显分化。就毒性频率分析,CYR34与G22-14毒性相当;CYR34与G22-14对某些单基因系鉴别寄主毒性更强,也存在毒性更弱的情况;Han等[27]在2015年评估了2种可侵染携带品种的小种对四川盆地小麦品种和育种系的毒力强弱,指出后来出现的小种(V26/贵22)比初始出现的小种(V26/CM42)毒性谱更宽,表明可侵染携带品种的小种已经进化为毒性更复杂的小种群体,结合G22-14快速增长的情况,可推断在病原菌与寄主植物的“军备竞争”过程中,G22-14获得了更强的毒性,使其在与群体中其他小种的竞争中取得生存优势,并在各地主栽品种的选择压力下发生了毒性分化;姚强等[28]用单基因系及SSR分子标记研究了CYR32及CYR33的分化,发现这两个流行小种在单基因系上出现了多样性分化,且不受地域的影响。本试验通过单基因系鉴别寄主鉴定所得优势小种race1的11株菌株中,通过中国鉴别寄主鉴定为CYR34的有3株,G22-14有2株,CYR33有1株,其余菌株均未通过中国鉴别寄主得到区分,更加说明中国鉴别寄主的区分力度不够,需要进行调整。上述结果表明,现有的这套中国鉴别寄主已不能反映当前中国条锈菌毒性变异的实际情况。
小麦条锈菌具有明显的毒性分化现象,利用鉴别寄主鉴定出不同的小种,可以较准确地反映小种群体与寄主群体之间的互作关系[1, 3, 29]。当前沿用的中国鉴别寄主主要由我国的历史重要生产品种和主要抗源品种组成,尽管能够明确某小种所能侵染的特定抗源品种类型,但由于这些品种抗锈基因尚不清楚,因此,无法推导小种所携带的无毒基因/毒性基因,也无法统计病原菌群体的毒性频率;此外,许多当前应用的已知抗性遗传背景的鉴别寄主携带有2个或2个以上的抗性基因,这些附加抗性基因可能限制了无毒基因与抗性基因互作结果的遗传学解释。实际上条锈菌自然群体小种毒性类型远高于报道的小种数量,对生产极具潜在威胁的新毒性小种未能及时监测和报道[30]。而单等基因系拥有相似遗传背景且抗性基因明确单一,在毒性小种频率动态监测领域更具优势[31]。因此,有必要筛选一套遗传背景相同、各鉴别寄主仅携带单个抗病基因并适合中国生产实际的单基因系(single gene lines)作为新的鉴别寄主体系,开展我国小麦条锈菌群体小种的系统精准鉴定。
4 结论
四川盆地与湖北南部群体同湖北西北部与河南南部群体存在遗传分化,明确冬繁区菌源不仅来自西北越夏区,而且来自西南越夏区;贵农22类群是冬繁区的最大流行类群;CYR34与G22-14的毒性分化证实生理小种内部存在明显分化,相较于中国鉴别寄主,单基因系鉴别寄主能够更精准地进行中国小麦条锈菌群体小种的鉴定。
[1] CHEN X M. Epidemiology and control of stripe rust [f. sp.] on wheat. Canadian Journal of Plant Pathology, 2005, 27(3): 314-337.
[2] WELLINGS C R. Global status of stripe rust: a review of historical and current threats. Euphytica, 2011, 179(1): 129-141.
[3] 康振生, 王晓杰, 赵杰, 汤春蕾, 黄丽丽. 小麦条锈菌致病性及其变异研究进展. 中国农业科学, 2015, 48(17): 3439-3453.
KANG Z S, WANG X J, ZHAO J, TANG C L, HUANG L L. Advances in research of pathogenicity and virulence variation of the wheat stripe rust fungusf. sp.. Scientia Agricultura Sinica, 2015, 48(17): 3439-3453. (in Chinese)
[4] 李振岐, 曾士迈. 中国小麦锈病. 北京: 中国农业出版社, 2002.
LI Z Q, ZENG S M. Wheat Rust in China. Beijing: China Agriculture Press, 2002. (in Chinese)
[5] CHEN W Q, WELLINGS C, CHEN X M, KANG Z S, LIU T G. Wheat stripe (yellow) rust caused byf. sp.Molecular Plant Pathology, 2014, 15(5): 433-446.
[6] 陈万权, 康振生, 马占鸿, 徐世昌, 金社林, 姜玉英. 中国小麦条锈病综合治理理论与实践. 中国农业科学, 2013, 46(20): 4254-4262.
CHEN W Q, KANG Z S, MA Z H, XU S C, JIN S L, JIANG Y Y. Integrated management of wheat stripe rust caused byf.sp.in China. Scientia Agricultura Sinica, 2013, 46(20): 4254-4262. (in Chinese)
[7] 黄冲, 姜玉英, 纪国强, 张国芝, 李辉, 李亚红. 2017年我国小麦条锈病流行大尺度时空动态分析. 植物保护学报, 2018, 45(1): 20-26.
HUANG C, JIANG Y Y, JI G Q, ZHANG G Z, LI H, LI Y H. Spatiotemporal dynamics of wheat stripe rust epidemics at regional level in China in 2017. Journal of Plant Protection, 2018, 45(1): 20-26. (in Chinese)
[8] HUANG L, YANG H, XIA C J, LI H F, WANG J F, WANG A L, ZHANG M, KANG X H, GAO L, ZHOU Y L, CHEN W Q, LIU T G. Long-distance transport off. sp.by upper airflow on the Yunnan-Guizhou Plateau disrupts the balance of agricultural ecology in central China. Plant Disease, 2022, 106(11): 2940-2947.
[9] ZHAN G M, JI F, CHEN X M, WANG J X, ZHANG D L, ZHAO J, ZENG Q D, YANG L J, HUANG L L, KANG Z S. Populations off. sp.in winter spore production regions spread from southwestern oversummering areas in China. Plant Disease, 2022, 106(11): 2856-2865.
[10] CHEN X M, MOORE M, MILUS E A, LONG D L, LINE R F, MARSHALL D, JACKSON L. Wheat stripe rust epidemics and races off. sp.in the United States in 2000. Plant Disease, 2002, 86(1): 39-46.
[11] ALJANABI S M, MARTINEZ I. Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic Acids Research, 1997, 25(22): 4692-4693.
[12] 孟岩, 杨彩柏, 姜舒畅, 黄丽丽, 康振生, 詹刚明. 基于KASP技术的小麦条锈菌SNP分子标记开发与评价. 植物保护学报, 2020, 47(9): 65-73.
MENG Y, YANG C B, JIANG S C, HUANG L L, KANG Z S, ZHAN G M. Development and evaluation of SNP molecular markers of wheat stripe rust based on KASP technology. Journal of Plant Protection, 2020, 47(9): 65-73. (in Chinese)
[13] KOSMAN E, LEONARD K J. Conceptual analysis of methods applied to assessment of diversity within and distance between populations with asexual or mixed mode of reproduction. The New Phytologist, 2007, 174(3):683-696.
[14] PRITCHARD J K, STEPHENS M, DONNELLY P. Inferenceof population structure using multilocus genotype data. Genetics, 2000, 155(2):945-959.
[15] JAKOBSSON M, ROSENBERG N A. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure.Bioinformatics, 2007, 23(14): 1801-1806.
[16] ROSENBERG N A. Distruct: a program for the graphical display of population structure. Molecular Ecology Notes, 2004, 4(1): 137-138.
[17] MANTEL N. The detection of disease clustering and a generalized regression approach. Cancer Research, 1967, 27(2): 209-220.
[18] AGAPOW P M, BURT A. Indices of multilocus linkage disequilibrium. Molecular Ecology Notes, 2001, 1(1/2): 101-102.
[19] WRIGHT S. Evolution in mendelian populations. Bulletin of Mathematical Biology, 1990, 52: 241-295.
[20] 曲若竹, 侯林, 吕红丽, 李海燕. 群体遗传结构中的基因流. 遗传, 2004, 26(3): 377-382.
QU R Z, HOU L, Lü H L, LI H Y. The gene flow of population genetic structure. Hereditas, 2004, 26(3): 377-382. (in Chinese)
[21] WANG H G, YANG X B, MA Z H. Long-distance spore transport of wheat stripe rust pathogen from Sichuan, Yunnan, and Guizhou in southwestern China. Plant Disease, 2010, 94(7): 873-880.
[22] LI M J, ZHANG Y H, CHEN W Q, DUAN X Y, LIU T G, JIA Q Z, CAO S Q, XU Z. Evidence for Yunnan as the major origin center of the dominant wheat fungal pathogenf. sp.. Australasian Plant Pathology, 2021, 50(2): 241-252.
[23] MCINTOSH R, MU J M, HAN D J, KANG Z S. Wheat stripe rust resistance gene/: A retrospective review. The Crop Journal, 2018, 6(4): 321-329.
[24] 黄瑾, 贾秋珍, 张勃, 孙振宇, 黄苗苗, 金社林. 小麦条锈病菌新菌系G22-9(CYR34)和G22-14流行趋势预测. 植物保护学报, 2018, 45(1): 101-108.
HUANG J, JIA Q Z, ZHANG B, SUN Z Y, HUANG M M, JIN S L. Epidemic forecasting of the new strains G22-9(CYR34) and G22-14 off. sp. tin wheat in Gansu province. Journal of Plant Protection, 2018,45(1): 101-108. (in Chinese)
[25] BAI B B, LIU T G, LIU B, GAO L, CHEN W Q. High relative parasitic fitness of G22 derivatives is associated with the epidemic potential of wheat stripe rust in China. Plant Disease, 2018, 102(3): 483-487.
[26] HUANG L, XIAO X Z, LIU B, GAO L, GONG G S, CHEN W Q, ZHANG M, LIU T G. Identification of stripe rust resistance genes in common wheat cultivars from the Huang-Huai-Hai region of China. Plant Disease, 2020, 104(6): 1763-1770.
[27] HAN D J, WANG Q L, CHEN X M, ZENG Q D, WU J H, XUE W B, ZHAN G M, HUANG L L, KANG Z S. Emerging-virulent races off.are threatening wheat production in the Sichuan basin, China. Plant Disease, 2015, 99(6): 754-760.
[28] 姚强, 王洁荣, 孟岩, 詹刚明, 黄丽丽, 康振生. 中国小麦条锈病菌CYR32和CYR33的毒性及基因型多样性. 植物保护学报, 2018, 45(1): 46-52.
YAO Q, WANG J R, MENG Y, ZHAN G M, HUANG L L, KANG Z S. Virulence and genotypic diversity of wheat stripe rust races CYR32 and CYR33 in China. Journal of Plant Protection, 2018, 45(1): 46-52. (in Chinese)
[29] WANG C C, JIANG B B, LIANG J M, LI L F, GU Y L, LI J T, LUO Y, MA Z H. Population genetic structures off. sp.in the Gansu-Ningxia region and Hubei province, China. Genes, 2021, 12(11): 1712.
[30] 刘太国, 王保通, 贾秋珍, 章振羽, 李强, 曹世勤, 彭云良, 金社林, 李明菊, 刘博, 高利, 胡小平, 陈万权. 2010-2011年度我国小麦条锈菌生理专化研究. 麦类作物学报, 2012, 32(3): 574-578.
LIU T G, WANG B T, JIA Q Z, ZHANG Z Y, LI Q, CAO S Q, PENG Y L, JIN S L, LI M J, LIU B, GAO L, HU X P, CHEN W Q. physiological specialization off. sp.in China during 2010-2011. Journal of Triticeae Crops, 2012, 32(3): 574-578. (in Chinese)
[31] 万安民. 小麦条锈菌鉴别寄主和小种命名现状. 植物病理学报, 2003, 33(6): 481-486.
WAN A M. Differentials and nomenclature of races ofWest f. sp.Eriksson. Acta Phytopathologica Sinica, 2003, 33(6): 481-486. (in Chinese)
Population Genetic Analysis ofin Main Winter-increasing Areas Based on Virulent Phenotypes and Genotypes
GAO XinPei, ZHAO Jun, LIU BoFan, Guo Yi,KANG ZhenSheng, ZHAN GangMing
College of Plant Protection, Northwest A & F University/State Key Laboratory of Crop Stress Biology for Arid Areas, Yangling 712100, Shaanxi
【Objective】To clarify the virulence structure and genetic diversity ofpopulations in the major winter-increasing areas of China, and to provide reference for the prevention and control ofand the rational layout of wheat resistance genes in the winter-increasing areas and the wheat production in Huang-huai-hai. 【Method】A total of 148isolates were collected and isolated from the major winter-increasing areas such as Sichuan Basin, Hubei and southern Henan, and the virulence phenotype was identified by using Chinese differentials and single-gene lines, and 17 pairs of KASP-SNP primers were used to mark the isolates and complete the genotype analysis. 【Result】Based on the Chinese differentials, 14 known races and 63 unknown pathotypes were identified, among which CYR34 (16.2%), G22-14 (12.2%), CYR32 (6.8%), CYR33 (5.4%) were the dominant races (pathotypes); based on the single-gene lines, 113 races (pathotypes) were identified, among which race1 (7.4%), race2 (3.4%), race3 (3.4%) were the dominant races (pathotypes). The Guinong 22 group was the largest epidemic group ofpopulation in China’s winter-increasing area, and all testedisolates did not infect single-gene lines varieties carryingand. The virulence phenotype and genotype of CYR34 and G22-14 showed diversification by single-gene lines virulence identification and molecular marker, indicating that there was high differentiation within these two dominant races. The clustering based on the virulence data of two sets of differentials showed that thepopulations in Sichuan Basin and southern Hubei were similar, while thepopulations in northwestern Hubei and southern Henan were similar; the genetic clustering based on KASP-SNP molecular data showed that there was genotype differentiation between thepopulations in Sichuan Basin, southern Hubei and northwestern Hubei, southern Henan; Structure analysis showed that Sichuan Basin, southern Hubei population mainly had two genetic backgrounds, northwestern Hubei, southern Henan population mainly had one genetic background; population genetic differentiation analysis showed that Sichuan Basinpopulation and southern Henanpopulation had the largestvalue, which was 0.118, with the largest genetic difference and obvious genetic differentiation; northwestern Hubei population and southern Henan population had the smallest degree of genetic differentiation,value was 0.010; gene flow analysis obtainedm value between northwestern Hubei population and southern Henan population was 25.236,m>4, there was a high-level gene flow between them, northwestern Hubei and southern Henan population and Sichuan Basin population hadm values of 2.923 and 1.864 respectively, both had a low-level gene flow; genetic diversity analysis results showed that Sichuan Basin, southern Hubei regionpopulation had a high-level of genetic diversity, northwestern Hubei, southern Henanpopulation had a low-level of genetic diversity. The above conclusions all support that Sichuan Basin, southern Hubei population has genetic differentiation with northwestern Hubei, southern Henan population. 【Conclusion】Single-gene lines can accurately identify Chineseraces;populations in China’s major winter-increasing areas have different sources.
wheat stripe rust; winter-increasing area; virulence identification; KASP-SNP; population genetics

10.3864/j.issn.0578-1752.2023.14.001
2023-02-27;
2023-04-11
国家重点研发计划(2021YFD1401000)、国家自然科学基金(32172380)
高新培,E-mail:1350261784@qq.com。赵鋆,E-mail:1138094105@qq.com。高新培和赵鋆为同等贡献作者。通信作者詹刚明,E-mail:zhangangming@nwsuaf.edu.cn。通信作者康振生,E-mail:kangzs@nwsuaf.edu.cn
(责任编辑 李莉)
