礼泉醪糟微生物多样性分析
2023-08-05党辉,陈佩
党 辉,陈 佩
(1.陕西师范大学食品工程与营养科学学院,陕西 西安 710119;2.陕西开放大学中瑞旅游与酒店管理学院,陕西 西安 710119)
醪糟又称“甜酒”,是一种以糯米为主要原料发酵而成的传统美食,其在我国陕西等地区自古就有酿造和食用的传统[1]。礼泉醪糟作为醪糟的典型代表,源于汉而盛于唐,其酿造历史悠久、口味香甜、酒香醇厚,被誉为“三秦第一家”[2]。醪糟在发酵基质和发酵条件上与米酒有异曲同工之妙,均可被称为“酒酿”或“甜酒”,常被进行比较研究[3]。值得注意的是,醪糟和米酒的制作过程均有微生物参与[4],且两者发酵过程和品质形成与微生物之间存在紧密的联系[5]。
近年来,国内越来越多的学者针对醪糟或米酒展开了一系列研究[6]。向凡舒等[7]利用湖北孝感和四川成都地方的米酒曲进行了米酒的发酵制作,并从米酒中分离鉴定出77 株乳酸菌,鉴定发现其主要隶属于片球菌属(Pediococcus)、乳杆菌属(Lactobacillus)和魏斯氏菌属(Weissella)等。王艳萍等[8]对荆州米酒中的微生物进行了分离纯化,发现毛霉属(Mucor)和酵母菌属(Saccharomyces)是米酒发酵过程中的优势菌属。但上述结论受限于纯培养的制约,仅能对环境样本中极少量的微生物进行鉴定[9]。而随着测序技术的发展和测序成本的迅速降低,人们越来越倾向于通过测序技术对传统发酵食品中微生物群落结构进行探究[10]。Wang Chunxiao等[11]通过测序技术发现不同米酒曲中的真菌构成存在较大差异,但整体来说均以根霉菌属(Rhizopus)和Saccharomyces为主;向凡舒等[12]通过对宣恩米酒中微生物多样性进行解析发现其主要以Pediococcus为主,其次依次是芽孢杆菌(Bacillus)和Lactobacillus等。礼泉醪糟作为西北地区一项传统的发酵美食,蕴含着丰富的微生物资源,但却少有研究对其微生物资源进行解析和收集,这可能导致微生物资源的大量丢失。
本研究以陕西咸阳礼泉的10 份农家自制醪糟为研究对象,使用高通量测序技术对其细菌和真菌多样性进行解析,并结合PICRUSt和BugBase等生物信息学软件对醪糟中细菌的功能和表型进行预测。通过本研究的开展,旨在为礼泉醪糟中微生物资源的搜集提供一定理论依据,并为礼泉醪糟的产业化提供技术支持。
1 材料与方法
1.1 材料与试剂
醪糟 陕西省咸阳市礼泉县的农户。
DNeasy mericon Food Kit DNA基因组提取试剂盒 德国QIAGEN公司;dNTPs Mix、5×TransStartTMFastPfuBuffer、FastPfuFly DNA Polymerase 北京全式金生物技术有限公司;DNA 1000试剂盒 美国Agilent公司。
1.2 仪器与设备
ND-2000C微量紫外分光光度计 美国Nano Drop公司;vetiri梯度基因扩增仪 美国AB公司;R950机架式服务器 美国Dell公司;MiSeq高通量测序平台 美国Illumina公司。
1.3 方法
1.3.1 醪糟样品的采集
从陕西省咸阳市礼泉县的农户家中共收集10 份农家自制的醪糟样本(编号LQ1~LQ10)。所有醪糟样本均以糯米为原料,接种自家自制酒曲,于28 ℃左右发酵2 d左右而成。采用无菌勺将醪糟搅拌均匀后挖取100 g左右装入无菌无酶的离心管中,封盖后置于低温保藏箱中运回实验室,于-80 ℃冰箱中贮存备用。
1.3.2 醪糟样品中宏基因组DNA的提取、扩增和测序
称取3 g左右醪糟样本于灭菌的研钵中,充分研磨至糊状。使用QIAGEN DNeasy mericon Food Kit DNA基因组提取试剂盒,对研磨后醪糟样本中微生物的宏基因组DNA进行提取,并使用微量紫外分光光度计对DNA浓度和纯度进行检验,将提取合格的DNA(OD260nm/OD280nm值在1.8~2.0)贮存至-20 ℃备用。
使用带有7 个碱基核苷酸标签(barcode)的ITS3F/ITS4R和338F/806R引物,参照文献中聚合酶链式反应(polymerase chain reaction,PCR)扩增条件和体系对真菌ITS区和细菌16S rRNA V3~V4区进行PCR扩增[13-14],使用微量紫外分光光度计和琼脂糖凝胶电泳对扩增产物的浓度和纯度进行检测。最后,将检验合格的扩增产物送至上海美吉生物医药科技有限公司,使用Illumina MiSeq PE300平台进行高通量测序。
1.3.3 生物信息学分析
根据双端序列的重叠区域对下机序列进行拼接和质控,并基于barcode信息将每条序列分配到不同样本中以得到高质量的测序数据集[15]。本研究基于QIIME(v1.75)平台对醪糟样本中微生物构成和多样性进行解析。首先,参照Yang Chengcong等[9]奶酪中细菌多样性的分析步骤对醪糟样本中细菌构成和多样性进行分析,接着参照王玉荣等[16]鲊广椒中真菌多样性的分析步骤对醪糟样本中真菌构成和多样性进行分析。本研究将不同数据库与代表性序列进行了同源性比对[17-18],通过Chao1指数和Shannon指数对醪糟中微生物的丰富度和多样性进行评估,并基于Bray-Curtis距离的主坐标分析(principal coordinate analysis,PCoA)对醪糟样品中微生物的整体结构进行分析。
依据Greengene数据库对高质量的16S rRNA序列进行聚类和注释,使用PICRUSt软件对礼泉醪糟样品中细菌的基因功能进行预测,并参照蛋白质直系同源簇数据库进行功能注释[19]。将对构建的操作分类单元(operational taxonomic units,OTU)和分组文件上传在线网站进行细菌表型预测(https://bugbase.cs.umn.edu/)[20]。
1.4 多元统计学分析
基于Bray-Curtis距离对醪糟样本中微生物群落的整体结构进行PCoA;基于Procrustes分析对醪糟样品中细菌和真菌的一致性进行研究;使用Wilcoxon test对α多样性指数、优势菌群、细菌功能和表型之间的差异性进行检验;对优势菌属之间的Spearman相关性进行分析。所用分析和可视化均使用R软件完成。
2 结果与分析
2.1 醪糟中微生物多样性分析
PCoA作为一种非约束性的数据降维方法,常被用来研究样本中微生物群落的差异性或相似性。本研究基于Bray-Curtis距离对醪糟中微生物差异性进行了分析,结果如图1所示。

图1 基于Bray-Curtis距离的PCoA(A)和Procrustes分析(B)Fig.1 Principal coordinate analysis (A) and Procrustes analysis (B) based on Bray-Curtis distance
由图1A可知,PC1的贡献率为40.75%,而PC2的贡献率为35.06%,两者累计贡献率为75.81%。由此可见,前2 个PC能够代表不同样本间群落结构的大部分差异。同时,不同农户家中自制的醪糟在空间分布上呈现出一定的分离和聚类趋势。因此,本研究以PC1为分组依据将10 份醪糟分为两组,其中LQ1、LQ2、LQ3、LQ9和LQ10隶属于A组,而其他样本则隶属于B组。通过Procrustes分析对醪糟样本中细菌和真菌的一致性进行分析发现(图1B),礼泉醪糟中的细菌群落结构与真菌群落结构之间不具有显著一致性(P=0.40)。由此说明,醪糟样本中的细菌和真菌可能相互影响较小。基于Bray-Curtis距离两组样品细菌和真菌群落结构的差异性分析如图2所示。
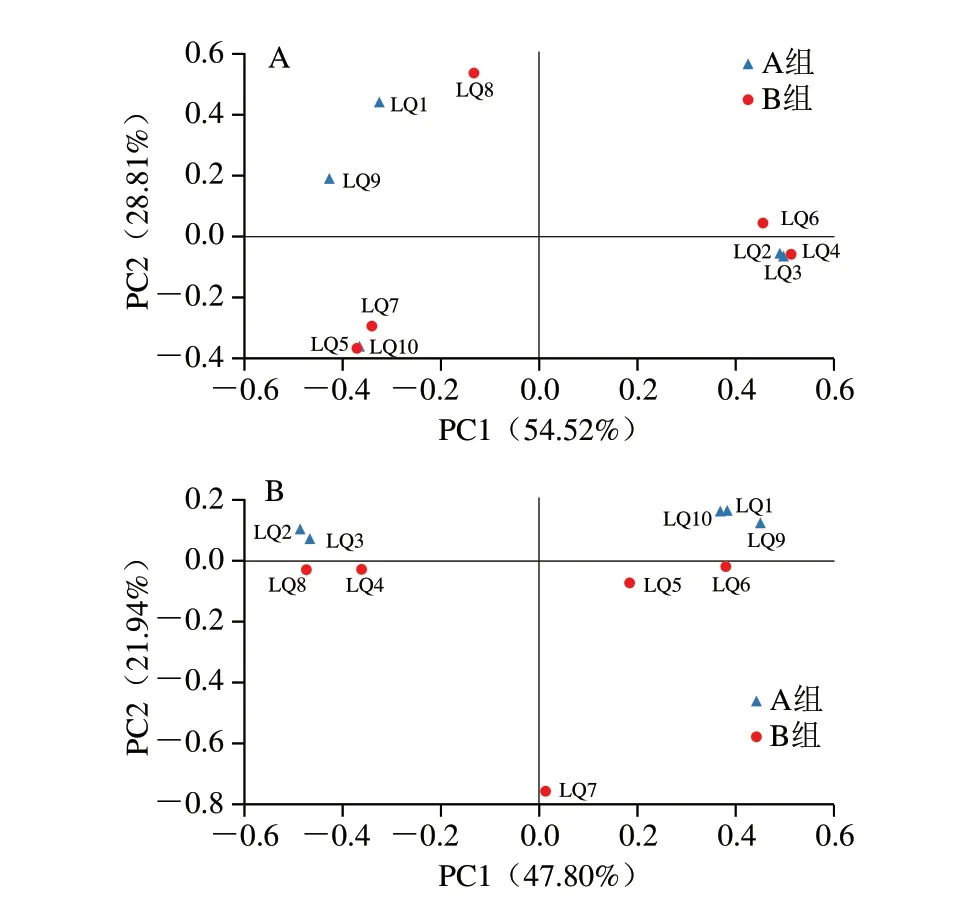
图2 基于Bray-Curtis距离细菌(A)和真菌(B)的PCoAFig.2 Principal coordinate analysis of bacterial (A) and fungal (B) communities based on Bray-Curtis distance
由图2A可知,隶属于A组的醪糟在4 个象限均有分布,而隶属于B组的醪糟样本则分布在第2、3和4象限,且隶属于两组的醪糟样本在空间上存在较大重合区域。由此可见,隶属于不同分组醪糟中细菌的群落结构无明显差异。由图2B可知,隶属于A组的醪糟全部位于Y轴正方向,而隶属于B组的醪糟则全部位于Y轴负方向,两组醪糟在空间上具有明显分离趋势,这也表明隶属于不同分组醪糟中真菌的群落结构存在明显差异。综上所述,真菌可能是造成醪糟中微生物群落整体结构差异较大的原因之一。在对醪糟中微生物群落结构进行解析的基础上,本研究进一步对不同分组中微生物的α多样性进行了比较分析,其α多样性指数的比较箱型图如图3所示。
由图3可知,A组醪糟中细菌的Chao1指数与B组无显著差异(P>0.05),而Shannon指数却显著高于B组(P<0.05);但A组醪糟中真菌的Chao1指数显著高于B组(P<0.05),而Shannon指数与B组无显著差异(P>0.05)。Chao1指数常被用来评估环境中微生物群落的丰富度,而Shannon指数则常被用来评估环境中微生物群落的均匀度。由此可见,A组醪糟中细菌的丰富度与B组无显著差异(P>0.05),但其细菌均匀度却显著高于B组(P<0.05);而在真菌上则呈现相反的趋势。
2.2 醪糟中微生物构成分析
在对醪糟中微生物多样性进行分析的基础上,本研究进一步对醪糟中优势菌门和优势菌属(平均相对丰度大于1%)进行了比较分析[16],其结果如表1所示。

表1 醪糟中优势菌门和菌属的比较分析Table 1 Comparative analysis of dominant microbial phyla and genera in Laozao
由表1可知,礼泉醪糟中的优势细菌门为厚壁菌门(Firmicutes)和变形菌门(Proteobacteria),平均相对丰度分别为97.20%和2.29%,其累计平均相对丰度高达99.49%;而优势细菌属有5 个,其分别为Lactobacillus(85.25%)、Pediococcus(5.50%)、明串珠菌属(Leuconostoc,1.68%)、链球菌属(Streptococcus,1.63%)和乳球菌属(Lactococcus,1.12%)。由表1 亦可知,礼泉醪糟中的优势真菌门为毛霉菌门(Mucoromycota)和子囊菌门(Ascomycota),平均相对含量分别为56.90%和43.10%,其累计平均相对丰度高达99.99%;而优势真菌属有4 个,其分别为Rhizopus(56.66%)、Saccharomyces(23.28%)、覆膜孢酵母属(Saccharomycopsis,12.24%)和假丝酵母属(Candida,5.35%)。经Wilcoxon检验发现,Pediococcus、Mucoromycota和Rhizopus在A组醪糟中的相对丰度显著高于B组,而Ascomycota则呈现相反的趋势。由此可见,醪糟中真菌的组间差异明显大于细菌。在此基础上,本研究进一步对醪糟中微生物的相关关系进行了分析,其结果如图4所示。

图4 醪糟中优势菌属的相关性分析Fig.4 Correlation analysis of dominant microbial genera in Laozao
由图4可知,醪糟样本中微生物之间相关性关系整体较弱,其中Lactobacillus和Saccharomycopsis,以及Leuconostoc和Lactococcus之间呈显著正相关(r>0.5,P<0.05),而Streptococcus和Saccharomycopsis之间呈显著负相关(r<-0.5,P<0.05)。而值得注意的是,细菌内部之间大多呈正相关,真菌内部之间无显著相关性,而细菌和真菌之间呈负相关。
2.3 功能和表型预测
随着生物信息学技术的快速发展,科研人员可以通过高通量测序数据对环境样本中细菌的功能和表型进行预测分析。本研究首先使用PICRUSt软件对醪糟中细菌的功能基因进行预测,并基于直系同源集(Clusters of Orthologous Groups,COG)数据库对其进行注释。醪糟中细菌功能类别的比较分析如图5所示。
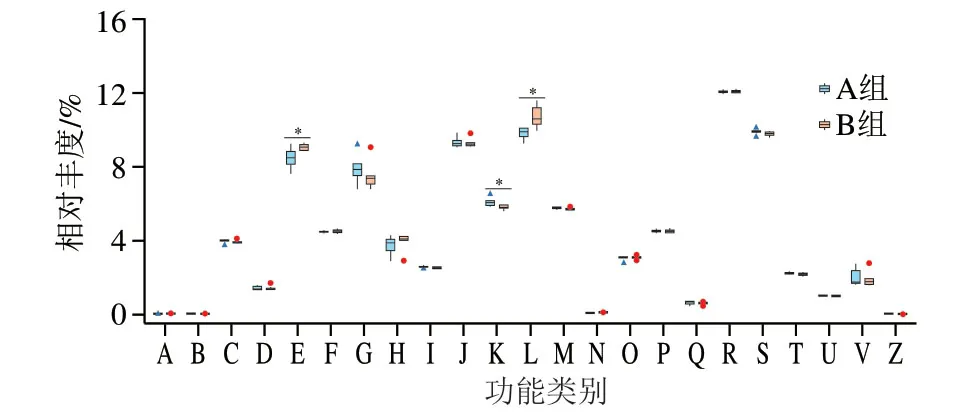
图5 醪糟中细菌功能类别的比较分析Fig.5 Comparative analysis of bacterial functional genes in Laozao
本研究从礼泉醪糟细菌中共注释到4021 个COG,其分别隶属于23 个一级功能层。由图5可知,一级功能层中氨基酸转运与代谢,复制、重组和修复在A组醪糟中相对表达量显著低于B组(P<0.05),而转录在A组醪糟中相对表达量显著低于B组(P<0.05)。而值得注意的是,翻译、核糖体结构与生物发生在醪糟中具有较高表达量。同时,本研究使用BugBase在线网站对醪糟中细菌的表型进行了预测,其结果如图6所示。
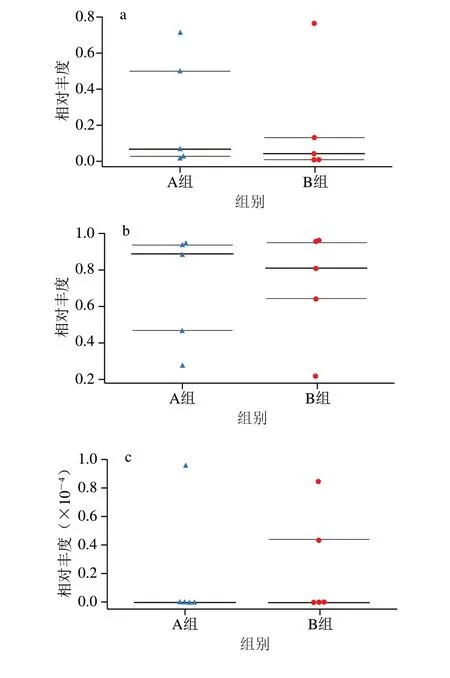

图6 醪糟中细菌表型结果的比较分析Fig.6 Comparative analysis of bacterial phenotypic data in Laozao
由图6可知,革兰氏阳性菌、革兰氏阴性菌、可移动元件和潜在致病性在两组中存在显著差异(P<0.05),其中革兰氏阴性菌和潜在致病性在A组中显著较高(P<0.05),而革兰氏阳性菌和可移动原件在A组中显著较低(P<0.05)。由此可见,A组和B组醪糟中细菌在表型上存在较大的差异。
讨论
近年来,随着食品工业的快速发展,越来越多的传统发酵食品消失在大众视野之中,而其中所蕴含的宝贵微生物资源也随之消失。受限于传统的微生物鉴定方法,在对传统发酵食品中微生物构成和多样性进行研究时,并不能准确揭示相关规律,这也使得相关人员无法准确解析传统发酵食品的发酵机制。因此,本研究采集了10 份礼泉醪糟,并结合高通量测序技术对其微生物群落结构进行解析,以期为其安全性评估和工业化生产提供必要的理论支持,促进我国相关食品产业的发展。
礼泉醪糟通常以糯米为主要原料酿制而成,因而其中可能蕴含着较为独特和多样的乳酸菌和酵母菌[21]。高通量测序结果显示礼泉醪糟中的细菌绝大部分隶属于Firmicutes,且优势细菌属为Lactobacillus、Pediococcus、Leuconostoc、Streptococcus和Lactococcus,累计占比高达95.18%,且优势菌属均隶属于乳酸菌。乳酸菌作为益生菌最大的来源库之一[22],其不仅能将糖类物质转化为乳酸,赋予发酵食品特殊的风味,抑制杂菌和有害菌的生长,且对于宿主的机体健康具有重要的意义[23]。王文平等[24]研究发现南宁地区米酒曲中以Lactobacillus(占比62.03%)为主,而孝感地区米酒中则以Weissella(占比50.14%)为主。上述结果均与本研究存在一定差异,这也说明礼泉醪糟具有较为独特的微生物群落结构,而制作环境可能是造成上述菌群差异的主要原因之一[25]。Lactobacillus作为醪糟发酵过程中的主要菌属,不仅将糖类物质转化为乳酸,其发酵过程还伴随着乙酸、乙醇和二氧化氮等副产物的产生,赋予礼泉醪糟特殊的口感和风味。大量研究证实了隶属于Lactobacillus的大量菌株在缓解机体炎症和调节肠道菌群平衡等方面发挥着巨大作用[26-27]。在对醪糟中真菌多样性进行研究时发现,其优势真菌属为Rhizopus、Saccharomyces、Saccharomycopsis和Candida,与大多数关于米酒中真菌多样性研究结果一致[28]。相关研究已经证实Rhizopus在米酒发酵过程中对淀粉水解起重要作用[29],而酵母菌在酒精产生中十分 重要[30]。由此可见,醪糟中微生物群落在醪糟发酵和品质形成过程中有重要意义。
通过对醪糟中微生物群落结构进行降维分析发现醪糟样本可明显划分为两个分组。进一步分析发现,两组中细菌的菌群结构无显著差异,但真菌的群落结构呈现出明显分离趋势,且通过对醪糟中优势菌群的差异分析验证了上述现象。由此可见,礼泉醪糟中细菌群落的整体结构较为相似,而真菌是造成微生物群落整体差异的主要因素。相关研究也证实了真菌可能在醪糟发酵过程中占据主导地位,而细菌则对于醪糟的品质形成有重要作用。通过基因功能预测发现A组中细菌具有较强氨基酸转运与代谢能力,这可能会导致醪糟的整体品质存在较大差异[31]。通过表型预测发现A组中细菌的潜在致病性相对较高,究其原因可能与农家醪糟的制作环境有关。综合采样信息发现,隶属于A组醪糟的制作环境较为开放,主要在阴暗的厨房中发酵而成,且发酵过程中较易受到各种杂菌和有害菌的污染。由此可见,改善传统发酵食品制作环境,对产品安全性和优良菌株筛选具有重要意义。
