同一家系中钼辅因子缺乏症相同MOCS1基因变异不同临床表型特点分析
2023-07-31关瑞莲赖莉明
关瑞莲 赖莉明
广州市妇女儿童医疗中心新生儿科(广州 510623)
钼辅因子缺乏症(molybdenum Cofactor deficiency,MoCD)是一种罕见且致命的遗传代谢性神经系统疾病,具有内源性中毒的病生理特点。发病率大约为1/20 万~1/10 万之间,发病的中位年龄是出生第1 天,诊断时的中位年龄为12.5 个月[1]。临床特点为早且快速进展的严重神经功能障碍;在症状出现初始阶段MRI 影像类似于缺氧缺血性脑病改变。因缺乏特征性表型极易漏诊误诊,确诊主要依据基因检测。MoCD 致病基因包括MOCS1、MOCS2、MOCS3 和GEPH,4 个基因对于MoCo 的合成都是必不可少的,任何一个基因的变异均可致一系列钼依赖酶活性丧失。特别是亚硫酸盐氧化酶的失活,将导致亚硫酸盐在脑部蓄积,抑制线粒体代谢,造成脑缺血性损伤。
MoCD 临床分为轻型和重型两种,前者表现温和,甚至可以参加正常教育,重型往往夭折于幼儿期。HINDERHOFER 等[2]根据全球已发表的具有临床和遗传数据的病 例(n=40),应用计算机技术建立基因型—表型相关性预测模型,试图对与MoCD 患者表型相关的基因型进行系统定量分析,以期能够进一步分类为严重或轻微。但局限于病例数确实偏少,需要更多的数据支持。
本文2 例患儿证实基因MOCS1 发生变异,是为新发突变(现有数据库未见收录),结合临床可以确诊为A 型MoCD。二者MOCS1 基因变异序列相同但临床表型特点却不相同,是否与表观遗传过程中基因组等的各种修饰有关,尚需进一步研究。本报告希望能对未来研究提供更多可靠的信息。
本研究已通过伦理委员会审核,编号:穗妇儿科伦批字【2022】第127A01 号。
1 资料与方法
1.1 病例1先证者,男性,第3 胎第3 产,胎龄37+3周,择期剖宫产,出生史无特殊,无窒息缺氧史,出生体重2 720 g。母孕期无特殊病史,父母非近亲结婚,智商正常,有1 健康正常姐姐(9 岁)。患儿生后出现呼吸困难予无创呼吸机辅助通气2 d,第2 天出现抽搐,同步床旁脑电图监测有电发作,首剂鲁米那15 mg/kg 止惊,后5 mg/(kg·d)维持,未再见惊厥发作。患儿无发热,各项感染指标、电解质、血气分析、脑脊液及尿有机酸、血浆氨基酸、酰基肉碱均未见异常。喂养困难,不会吸吮,持续吞咽康复训练3~4 周后逐渐能够自吮奶;出生1 周行头颅MRI 平扫+增强:双侧基底节肿胀,左侧基底节软化灶及出血,考虑缺氧缺血性脑损伤(图1A-B);MRA 未见异常改变;经颅多普勒显示颅内各血管未见异常;脑电图提示清醒及睡眠各期双侧额、中央、颞-多量棘尖波散发,左右不同步。生后1 个月复查头颅MRI 显示双侧基底节软化灶及出血继续加重,两侧脑室扩张(图1C-D);磁共振波谱分析(MRS)胆碱(Cho)峰值升高,N-乙酰天门冬氨酸(NAA)峰值下降(图2)。脑干听觉脑干诱发电位:双侧听神经传导通路损伤30 dB(左侧),40 dB(右侧);眼底检查未见晶状体脱位等异常表现;血尿酸、尿尿酸及生物蝶呤偏低。3 个月随访头颅MRI 示双侧基底节软化及出血较前吸收,脑室扩张与前相仿;无皮层下坏死或囊肿,无明显脑萎缩;双侧听神经传导通路30 dB(左侧),30 dB(右侧),V 波潜伏期延长较前好转;脑电图轻度异常:左侧额、中央区多量慢波及偶发尖波。复查血尿酸仍偏低,尿尿酸正常。患儿大运动发育稍差,但精神运动发育与同龄儿相仿;典型面部特征突出(图3)。
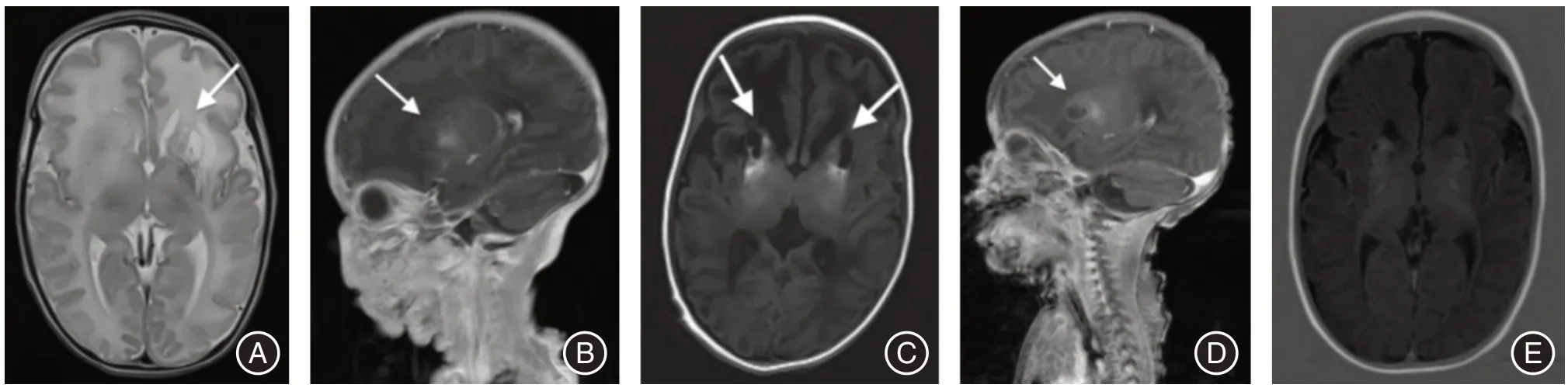
图1 先证者MRI 影像Fig.1 MRI imaging of proband

图3 先证者(3 个月)面容(已获得家长同意书)Fig.3 The face of the proband(3 m)(with parental consent)
1.2 病例2为先证者姐姐,其母第2 胎,女性,生后10 个月之前神经发育描述正常。10 月龄感冒后出现抽搐、四肢肌张力增高、继而进食困难。头颅MRI 显示双侧基底节肿胀明显,以双侧尾状核和豆状核为著,脑沟明显加深。脑干听觉脑干诱发电位:双侧听神经传导通路异常50 dB(左侧),70 dB(右侧)。动态复查头颅MRI 可见基底节病变持续加重和脑萎缩表现(图4)。因当时对该病认识的局限性,临床上以“病毒性脑炎”对症治疗,无明显疗效。神经发育显著倒退,迅速进展为不能独坐、不能翻身、表情呆滞等症状。现6 岁仍然存活,呈全面脑瘫表现,消瘦,营养欠佳,流质喂养。

图4 先证者姐姐MRI 影像Fig.4 MRI images of the proband's sister
1.3 基因组DNA 提取和基因测序考虑到二者相类似的基底节病变,征得父母知情同意后,对患儿及其父母、第一、二胎进行全外显子测序,采用QIAamp DNA Blood Mini Kit(Qiagen,Hilden,Germany)提取基因组DNA 5 ng,用Ion-TorrentPGM 技术进行全外显子测序,人群频率数据库参考dbSNP、1000 Genomes、gnomAD、和HGMD,对检测到的所有变异位点进行生物信息学有害性预测分析,变异位点致病性分类参照遗传变异分类标准与指南对变异位点进行解读和致病性分类[3]。
2 结果
经全外显子测序检测结果发现患儿MOSC1 基因存在c.1275delA(p.Gly426Aspfs Ter10)位点的纯合突变,该变异为移码变异,在dbSNP 数据库及1000 Genomes 数据库中未见收录。在gnomAD 数据库中东亚人群频率为0.0001631(3 个杂合子),该基因在ClinVar 数据库收录为致病性/可能致病性的变异包括:移码变异8 个,错义变异1 个,无义变异6 个,经典剪接变异4 个,均与本例突变不同。综合患儿临床表现、影像学、生化检查及家系分析,证实为A 型MoCD,变异遗传自父母,父母为杂合突变(图5)。
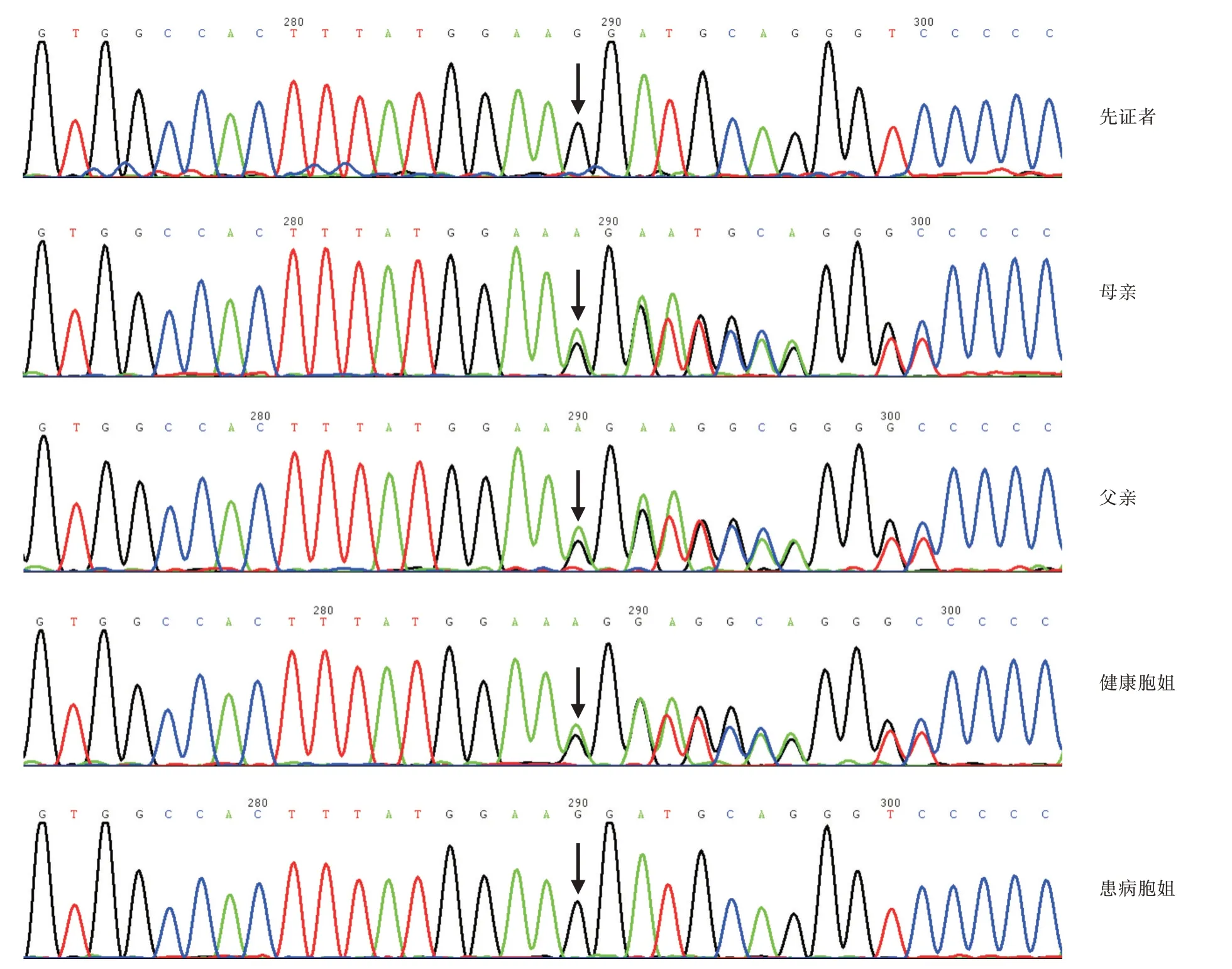
图5 家系MOCS1 基因Sanger 测序图Fig.5 Sanger sequencing of MOCS1 gene in a family
3 讨论
为了更好地理解该病临床特点,首先要了解疾病发生的病生理过程。钼与独特的蝶呤结合形成钼辅因子,从而获得催化活性。MoCo 生物合成途径可分为3 个步骤[4]:MOCS1 将鸟苷三磷酸合成环状吡喃蝶呤单磷酸盐(cPMP),MOCS2 和MOCS3将cPMP 转化为钼蝶呤,GEPH 插入钼形成钼辅因子。MoCD 是由于参与钼辅因子生物合成的4 个基因(MOCS1、MOCS2、MOCS3 和GEPH)变异所致,其中大约2/3 的MoCD 患者为MOCS1 基因突变,导致无法合成生物合成途径中的第一个中间体:环吡喃蝶呤单磷酸盐(cPMP),被归类为A 型;其次为MOCS2、MOCS3 基因突变,导致cPMP 无法转化为钼多菌素,被归类为B 型MoCD,GEPH 基因突变极罕见,归类为C 型MOCD。生物合成的任何步骤的缺陷都会导致依赖于MoCo 的酶(如亚硫酸盐氧化酶、黄嘌呤氧化还原酶、醛氧化酶)失活和线粒体α 胺肟还原组分的缺乏。部分患者有特殊面容,ARICAN 等[4]回顾性研究中35 例患者中有14 例出现小头畸形、双颊饱满、眼裂狭长、宽鼻梁、长人中等;本研究先证者外貌特征与近期中国MOCS1 案例报道较近似[5](图5)。
MoCD 使得亚硫酸盐氧化酶不能将亚硫酸盐转化为硫酸盐,导致亚硫酸盐蓄积,受累最明显的区域是基底节。亚硫酸盐在基底节的持续累积,是造成神经功能障碍的主要原因;而黄嘌呤脱氢酶活性丧失致嘌呤代谢障碍,但对神经系统无明显影响,醛氧化酶活性丧失多无明显临床表现[6]。因此MoCD 的表型效应主要局限于中枢神经系统,严重的神经系统损害目前认为是一系列代谢紊乱导致亚硫酸盐、牛磺酸、S-硫代半胱氨酸和硫代硫酸盐的积累,尤其是亚硫酸盐神经毒性水平的蓄积,致线粒体呼吸链异常,抑制了生物能量代谢,使大脑更易遭受缺血性损伤。典型的临床症状在新生儿早期就出现,以难治性癫痫发作及喂养困难最常见,通常神经障碍进展迅速。所有患儿均有运动发育落后、语言发育落后;本研究先证者生后即出现较典型临床症状,是为早发型,说明代谢障碍导致的神经毒性在宫内即已发生。但母孕期无特殊疾病,胎儿宫内发育正常,定期产检超声未见脑室扩张等异常表现而未予重视。值得注意的是先证者虽为早发,但后期临床表型温和,脑损伤似乎停止,生后1 个月、3 个月龄随访MRI 显示基底节损伤逐渐修复,神经心理发育评估稍落后;生后不久给予吸吮吞咽康复训练后,逐渐自吮奶,营养发育正常。而迟发型MoCD 患者与经典型不同,通常在2 岁内发病,往往在发生疾病后出现神经障碍表现,癫痫发作不常见[7],临床表型亦各不相同,既有迅速进展的神经功能恶化表现,亦有轻型的温和表型。先证者患病胞姐生后未见明显症状,10 个月因“发热”起病,神经障碍进展迅速,目前已出现全面神经心理发育落后、痉挛性瘫痪。
亦有报道同一家族的相同突变出现不同的临床病程[8]。本研究的2 例患儿发病时间及神经障碍程度均显著不同。第2 例为迟发型,因“发热”出现神经障碍表现,随后神经功能恶化迅速;而第1 例虽为典型早发型,但是神经障碍进展不明显,呈现温和的临床表现,无持续反复的癫痫发作,2 例患儿未体现明显基因-表型相关性。基因型和表型之间密切相关,基因型直接决定了表型的形成,但是基因含量不一定完全决定表型的表现。因为表观遗传过程还受其他因素影响,比如基因组DNA 甲基化修饰、组蛋白修饰、非编码RNA 调控、染色质重塑等[9]。因此有可能在基因组DNA序列没有发生改变的情况下,基因功能发生了可遗传的变化,并最终导致了表型的变化。MAYR 等[10]对一名具有异常晚期发病和轻度表型的患者转录本进行生物信息学分析,发现突变位点下游的潜在Kozak 序列提供了独立表达MOCS1B 蛋白的可能性,这种微量功能的MOCS1B 蛋白足以保护患者免受MoCD 出现严重症状。
尽管涉及临床表现的其他因素的作用仍不清楚,但可以肯定疾病诱发是神经系统恶化的外部触发因素(如本研究病例2)。
MoCD 早期影像学检查需与新生儿缺氧缺血性脑病(neonatal hypoxic ischemic encephalopathy,HIE)鉴别,HIE 患者的影像学通常在分娩后1~2 周内稳定,而MoCD 患者的稳定性没有改善。本研究先证者没有围产期不良病史,生后很快出现神经系统症状,6 d 时MRI 提示基底节缺血性坏死(图2),而28 d 时MRI 显示病灶继续进展。MoCD患者早期脑水肿,白质和灰质受累,快速进展为皮层下坏死,囊性脑软化。基底节和齿状核改变通常被认为是迟发和轻度临床病程患者的MRI 中的独立表现,在HIE 中尾状核很少受到影响,而在MoCD 中总是受到影响。一些MRI 数据已用于产前诊断[11]。另一方面其核磁波谱分析Cho 峰通常升高,HIE 则相反[12]。本例先证者MRI 氢谱分析Cho 峰升高,NAA 峰下降,提示病变区神经元坏死、无氧代谢及胶质增生(图3)。
特异性生化检查是诊断MoCD 的重要辅助手段,为早期诊断提供一定线索;典型的MoCD 生化检查尿酸水平明显下降,尿嘌呤代谢物即黄嘌呤和次黄嘌呤增高;尿亚硫酸试验阳性,但该检测有一定的局限性,且大多数机构无法进行亚硫酸盐检测,且检测结果具有一定的假阴性。血尿酸也可呈正常表现[13],对诊断造成误导,尤其是症状较轻的患者。本研究的先证者及其姐姐尿酸及生物蝶呤降低。
A 型MoCD患者发病根本原因是体内无法合成环吡喃单磷酸(cyclic pyranopterin monophosphate,cPMP),2015 年一项多中心前瞻性队列研究已经证实,cPMP早期替代治疗对于A型MoCD有效且具有良好的安全性[14]。美国FDA 已经于2021 年2 月26 日批准Origin 生物科学公司研发的新药Nulibry(fosdenopterin)静脉注射剂用于治疗A 型钼辅因子缺乏症[15]。近期日本学者[16]报道了1 例晚期发病的轻型的患者,在1 岁4 月龄时接受低蛋白饮食,蛋白质限制3 个月后,尿亚硫酸盐试验变为阴性,尿S-磺基半胱氨酸水平下降,肌张力运动障碍得到改善,患者病程没有进展和癫痫发作。这一发现提示,饮食蛋白质限制有可能抑制轻型MoCD疾病进展。
本研究局限性在于MOCS1 基因是新发变异序列,临床特点既往没有报道,该变异序列是否存在钼辅因子依赖酶的残留活性需要更多研究证实;2 例出现不同表型特点如果从表观遗传学角度分析,比如转录水平测序、蛋白质翻译等方面继续做深入研究,可能会有新的发现。
终上所述,MoCD 是一种先天致命性常染色体隐性遗传代谢病,对于新生儿早期出现严重基底节损伤需与重度HIE 相鉴别,必要时完成基因检测。既然A 型MoCD 用cPMP 替代治疗已经可用,早期诊断和遗传咨询非常重要。由于数据稀缺,目前可识别的变异序列有限,尚需要更多有价值的遗传信息,建立有效诊断模型,尽早开始治疗,避免更严重后遗症发生。
