UPLC测定不同产地茯苓中5种三萜类成分的含量*
2023-07-27张丽君
张丽君,王 凯,刘 鸣,雷 丽,高 波,谭 沛,高 巍,3
(1 安徽华润金蟾药业股份有限公司,安徽 淮北 235000;2 华润三九医药股份有限公司,广东 深圳 518110;3 华润三九(六安)中药材产业发展有限公司,安徽 六安 237000;4 华润三九现代中药制药有限公司,广东 惠州 516000)
茯苓是我国大宗常用菌类药材,已有二千多年应用历史,具有利水渗湿,健脾宁心的功效[1-2],广泛用于临床配方。茯苓在我国广有分布,但经年采收,野生资源已稀存,药材主要依靠栽培提供,且不同产地栽培的茯苓药材存在一定的差异。2020版《中国药典》茯苓项下未制定含量测定项,不能对茯苓质量进行全面控制。以往文献研究显示,多糖和三萜类化合物为茯苓的主要化学成分为[3-4]。根据国内外报道,茯苓菌丝体和菌核中分离到的三萜类化合物多属于四环三萜类,且主要为羊毛甾型四环三萜[5]。本实验主要采用UPLC法,对茯苓中5种三萜类成分去氢土莫酸、猪苓酸C、3-表去氢土莫酸、去氢茯苓酸、松苓新酸[6-12]同时测定,以期对茯苓药材不同产地[13-14]的质量评价提供一定的参考。
1 仪器与试药
Thermo Vanquish超高效液相色谱仪,赛默飞科技有限公司;KQ-800 DE超声波清洗器,昆山市超声仪器有限公司;XPE 26、MS204S电子天平,METTLER TOLEDO公司。
12批茯苓药材分别从湖南、湖北、云南、安徽4个主产区收集,经安徽中医药大学刘守金教授鉴定为多孔菌科真菌茯苓Poriacocos(Schw.)Wolf 的干燥菌核、去氢土莫酸对照品(批号:170112-202012,纯度98.0%)、猪苓酸C对照品(批号:260087-202107,纯度96.0%)、松苓新酸对照品(批号:190166-202009,纯度98.0%),上海鸿永生物科技有限公司;去氢茯苓酸对照品(批号:21060701,纯度99.5%),成都格力普生物科技有限公司;3-表去氢土莫酸对照品(批号:020177-202107,纯度96.0%),中国食品药品检定研究院;乙腈为色谱纯;水为娃哈哈水;其它试剂均为分析纯。
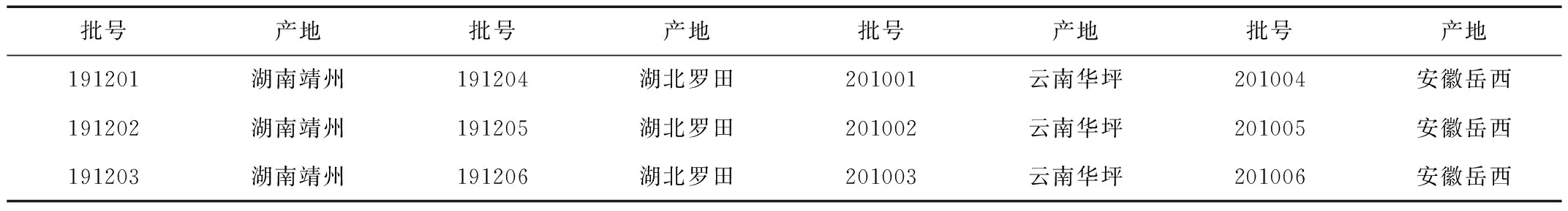
表1 茯苓样品来源信息
2 方法与结果
2.1 色谱条件
色谱柱:HSS T3-C18(2.1 mm × 100 mm,1.8 μm);流动相:乙腈-0.1%磷酸(78∶22);柱温:25 ℃;流速:0.3 mL/min;检测波长:243 nm;进样量:2 μL。
2.2 对照品溶液的制备
精密称取去氢土莫酸、猪苓酸C、3-表去氢土莫酸、去氢茯苓酸、松苓新酸对照品适量,加甲醇制成每1 mL中含去氢土莫酸25 μg、猪苓酸C 12.5 μg、3-表去氢土莫酸7.5 μg、去氢茯苓酸15 μg、松苓新酸5 μg的混合溶液,即得混合对照品溶液。
2.3 供试品溶液的制备
精密称定茯苓药材粉末(过三号筛)约1.0 g,置具塞锥形瓶中,加入甲醇10 mL,称定重量,超声提取30 min,放至室温,再称定重量,用甲醇补足损失的重量,滤过,即得。
UPLC色谱图见图1。
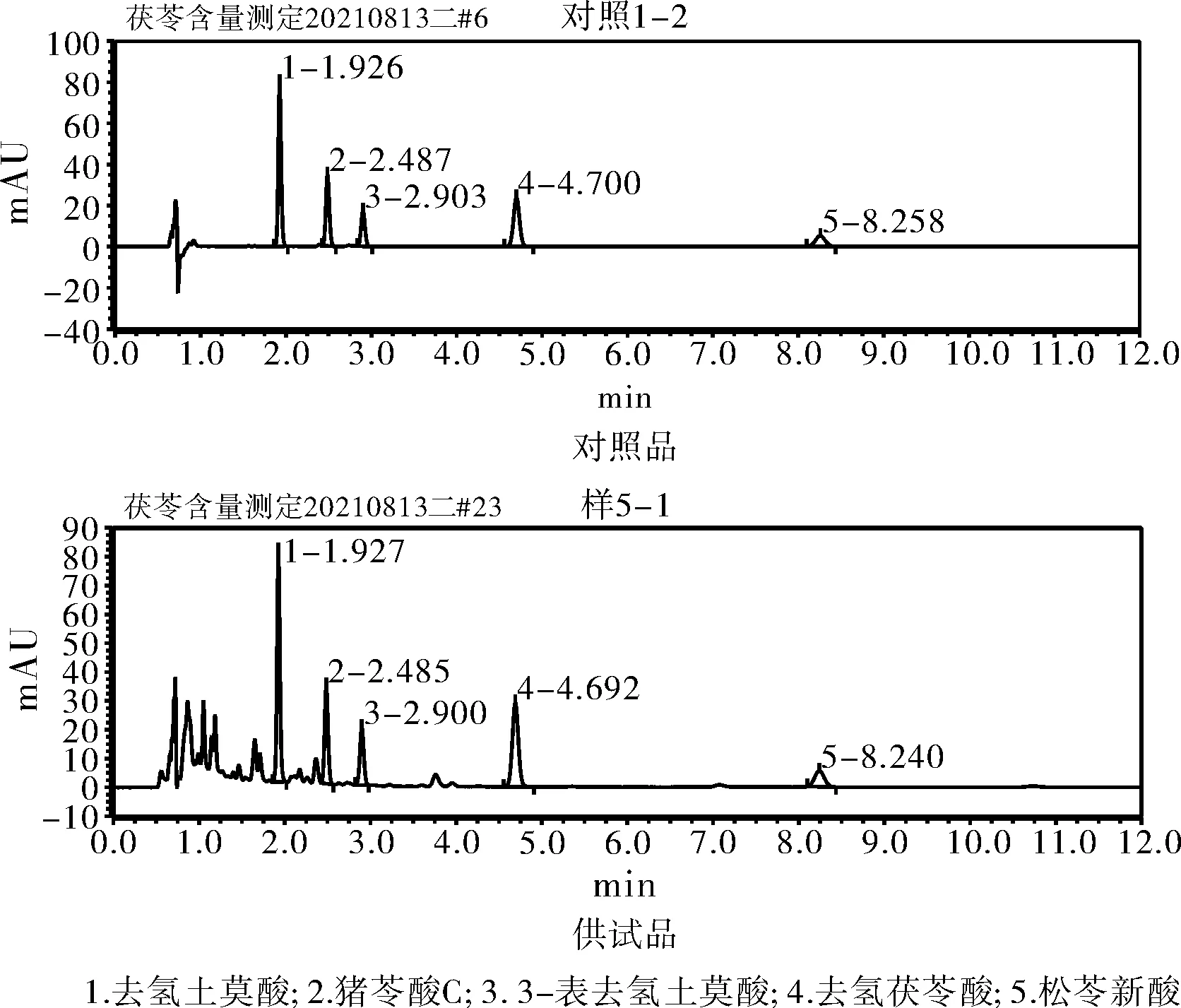
图1 茯苓UPLC
2.4 线性范围考察
精密称定5种三萜类指标成分对照品适量,加甲醇制成每1 mL中含去氢土莫酸0.1947 mg、猪苓酸C 0.0964 mg、3-表去氢土莫酸0.0548 mg、去氢茯苓酸0.1145 mg、松苓新酸0.0368 mg的混合对照品母液,精密吸取母液5 mL加甲醇定容至10 mL,依次等比稀释,得到7份系列浓度的混合对照品溶液,进样测定并记录相应峰面积。将峰面积做为纵坐标Y,将对照品进样量(μg)做为横坐标X,并进行线性回归,结果见表2。

表2 线性回归关系结果
2.5 供试品精密度试验
对同一份茯苓药材粉末(批号201004)精密称定,按供试品制备方法处理,同一供试品溶液连续进样6次,测定各指标成分的峰面积,结果5种三萜类指标成分的峰面积RSD均<2%,说明该方法仪器精密度良好。
2.6 重复性试验
对同一批茯苓药材粉末(批号201004)精密称定,按供试品制备方法处理,并进样测定,根据各指标成分峰面积计算含量质量分数的RSD,结果去氢土莫酸RSD为0.92%;猪苓酸CRSD为0.99%;3-表去氢土莫酸RSD为1.01%;去氢茯苓酸RSD为0.98%;松苓新酸RSD为0.92%,表明该方法具有良好的重复性。
2.7 稳定性试验
对同一份茯苓药材粉末(批号201004)精密称定,按供试品制备方法处理,并分别在第0,2,4,6,8,10,12 h进样测定,并计算各指标成分的峰面积RSD,结果去氢土莫酸RSD为0.10%;猪苓酸CRSD为0.09%;3-表去氢土莫酸RSD为0.19%;去氢茯苓酸RSD为0.19%;松苓新酸RSD为1.86%,表明供试品溶液在12 h内(样品盘25 ℃)稳定,可以满足测定需要。
2.8 加样回收率试验
对同一批已知含量的茯苓药材粉末(批号201004)6份(未扣水分去氢土莫酸、猪苓酸C、3-表去氢土莫酸、去氢茯苓酸、松苓新酸的含量分别为0.29、0.12、0.08、0.17、0.05 mg/g)精密称定,每份约0.5 g,另用甲醇配制每1 mL中含去氢土莫酸140 μg、猪苓酸C 65 μg、3-表去氢土莫酸42 μg、去氢茯苓酸87 μg、松苓新酸28 μg的混合对照品溶液,精密加入1 mL该混合对照品溶液至每份供试品样品中,再按2.3项下制备方法处理样品,并对含量进行测定,结果表明该方法回收率良好。计算结果见表3~表7。

表3 去氢土莫酸加样回收率
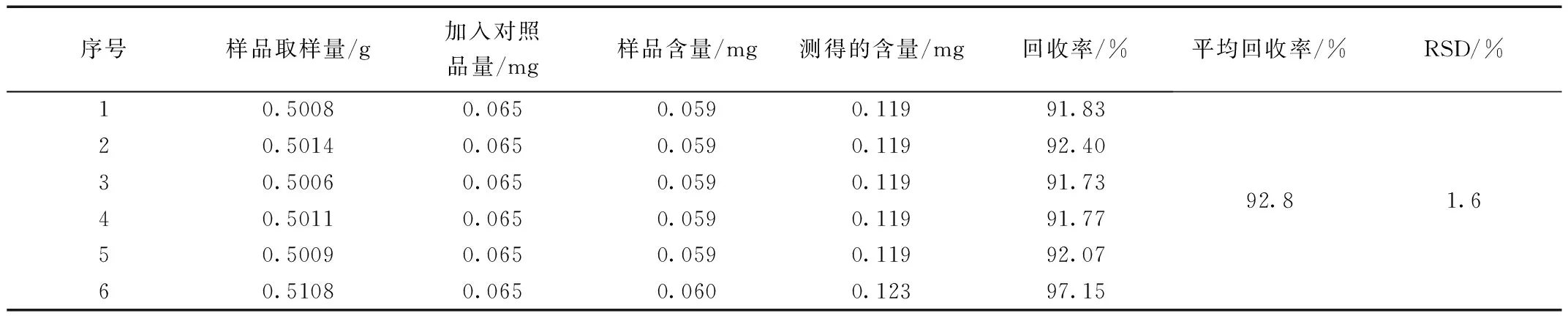
表4 猪苓酸C加样回收率

表5 3-表去氢土莫酸加样回收率
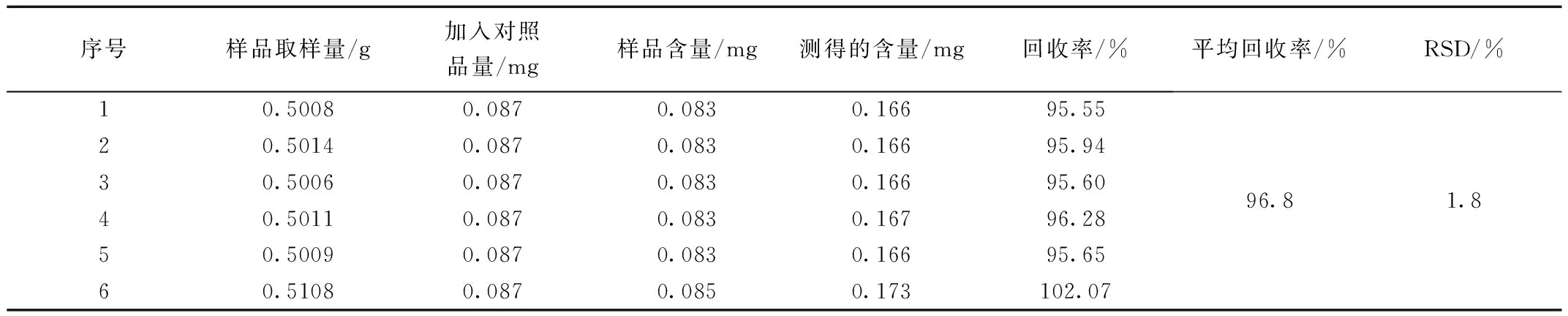
表6 去氢茯苓酸加样回收率

表7 松苓新酸加样回收率
2.9 样品测定
分别对12批不同产地的茯苓药材样品,按确定的供试品制备方法和色谱条件测定,5种三萜类成分的含量见表8。

表8 5种成分含量测定结果
3 讨 论
3.1 波长的选择
对去氢土莫酸等五种三萜类成分在190~400 nm下进行3D图谱全波长扫描,结果在波长为243 nm时五种三萜类目标成分均呈现出最大吸收,故最终确定茯苓药材样品的检测波长为243 nm。
3.2 流动相的选择
分别考察了乙腈-水,甲醇-水,乙腈-0.1%磷酸,甲醇-0.1%磷酸四种不同流动相,结果显示当流动相为乙腈-0.1%磷酸时,5种三萜类目标成分出峰时间较短,峰形、分离度等系统适应性参数也较好。
3.3 提取方法的选择
对供试品提取方法进行考察时,主要研究了超声和回流两种不同提取方法的差异,结果显示,当提取方法为超声时,5种三萜类目标成分色谱峰峰面积均高于回流提取,且超声在实际实验操作中更简便,故最终选择提取方法为超声;并进一步对不同提取溶剂(水、30%甲醇、50%甲醇、70%甲醇、甲醇、50%乙醇、乙醇)进行考察,对比试验数据结果发现,甲醇的提取效果更加充分完全;在此基础上,比较了甲醇超声不同提取时间(15 min、30 min、45 min)的差别,结果超声30 min、45 min各色谱峰峰面积基本无变化,故确定30 min为最终超声提取时间;进而考察了甲醇提取溶剂用量5 mL、10 mL、20 mL的提取效果,结果三者差异不大,考虑到操作性,最终选择提取溶剂用量为10 mL。
3.4 质量分析
4个产地茯苓药材中5种三萜类成分总含量差异不大,可能原因为产地样品的代表性不够,后续建议扩大收集样本量,进一步探讨各产地之间茯苓质量的差异。
4 结 论
三萜类化合物作为茯苓的主要有效成分,具有抗炎、利尿、免疫抑制、神经生长促进、抗癌等作用。本实验建立的UPLC方法可同时测定5种三萜类成分(去氢土莫酸、猪苓酸C、3-表去氢土莫酸、去氢茯苓酸、松苓新酸)含量,具有方法准确可靠、快速、操作性强等优点,能够为不同产地茯苓的质量控制提供一定的参考。
