LIPI-4 EII对单核细胞增生性李斯特菌关键毒力 因子转录调控的影响
2023-07-15史唯地刘彩霞寇丽君吕双飞任慧杰曾东东孔翠莲高盛杰钱瑞宣
史唯地 马 勋 刘彩霞 寇丽君 吕双飞 任慧杰 曾东东 王 静 孔翠莲 高盛杰 钱瑞宣
(石河子大学 动物科技学院,新疆 石河子 832000)
单核细胞增生性李斯特菌(Listeriamonocytogenes,LM)是引起人类与多种动物李斯特菌病的食源性人畜共患病原体,可穿越肠道屏障,血脑屏障和胎盘屏障[1-2]。李斯特菌病主要影响孕妇、新生儿、老年人和免疫缺陷者,并且呈现为危及生命的疾病,如母婴李斯特菌病、败血症和脑膜脑炎[1-2]。每年由已知食源性病原体引起的死亡中有28%是由李斯特菌引起的[3],且LM引起的食物中毒导致患者死亡率高达30%[4]。目前WHO将其列为21世纪四大食源性致病菌之一,对公共卫生安全造成了较大危害[5]。
与LM致病力密切相关的毒力岛(Listeriapathogenicity island, LIPI)包括LIPI-1、LIPI-2、LIPI-3和LIPI-4。其中LIPI-1与LM的胞内感染相关,主要由prfA、plcA、hly、mpl、actA和plcB6个毒力基因组成[6]。LIPI-2与LM的黏附、侵袭有关,由inlA、inlB和inlC等多个内化素基因组成[7]。LIPI-3可编码李斯特菌溶血素S,与LM的细胞毒性和溶血性相关[8]。2016年 Maury等[9]发现LIPI-4是一个高毒力基因簇,最先被认为是CC4克隆群的LIPI-4为第一个与神经系统感染和母胎感染特异性关联的LM毒力因子,但是近两年来发现ST87和ST213等非CC4克隆群分离株都含有LIPI-4的特异基因[10-11]。该毒力岛包含6个基因,分别编码麦芽糖-6′-磷酸葡萄糖苷酶、抗转录终止子、与PTS系统相关的未知蛋白、EIIA、EIIB和EIIC[12-13],其本质上是一个纤维二糖家族磷酸烯醇丙酮酸(Phosphoenolpyruvate,PEP)磷酸转移酶系统(Phosphotransferase system,PTS)。有研究表明,PTS糖特异性组分EII不仅能够响应糖的运输,还影响细菌毒力基因的表达[14-16]。Aké等[17]发现LM甘露糖-PTS的EIIAB缺失株毒力基因转录水平的上调依赖于PrfA的表达,同时PrfA的活性和毒力基因表达与EII的去磷酸化的水平负相关。因此,研究LIPI-4EII以揭示LM的致病机制是很有必要的。阮婷玉等[18]研究表明LIPI-4的缺失大大降低了对永生化人脑微血管内皮细胞(Human cerebral microvascular endothelial cells,HCMEC/D3)的侵袭和胞间传播能力,进而影响LM的毒力。而LIPI-4作为LM中最新发现的毒力岛,EII在LIPI-4参与LM致病性机制中发挥的作用尚不清楚。
本研究通过同源重组技术构建LIPI-4中EII(EIIA、EIIB和EIIC)的缺失株、强启动子回补株和天然启动子回补株,研究其对关键毒力因子转录水平及胞内增殖能力的影响,以此来研究EII对LM毒力因子的调控作用,为深入解析LIPI-4在LM感染宿主过程中的机制奠定基础。
1 材料与方法
1.1 菌株、质粒和细胞
LM928由本实验室分离、鉴定及保存;大肠杆菌DH5α感受态细胞购自北京全式金生物技术有限公司;pMD19-T(simple)质粒载体购自大连TaKaRa公司;温度敏感型穿梭质粒pKSV7由浙江大学动物科学学院提供;整合型质粒pIMK2由扬州大学生物科学与技术学院殷月兰教授惠赠;永生化人脑微血管内皮细胞(HCMEC/D3)购自北京北纳创联生物技术研究院。
1.2 培养基与主要试剂
脑心浸出液培养基(BHI)和LB培养基购自青岛高科园海博生物技术有限公司;氨苄青霉素、氯霉素、卡那霉素和庆大霉素购自北京博奥拓达科技有限公司;限制性核酸内切酶(PstⅠ、SacⅠ和XhoⅠ)、高保真DNA聚合酶和T4 DNA连接酶购自大连TaKaRa公司;DMEM培养基、胎牛血清(FBS)、胰蛋白酶和青链霉素(P/S)购自美国 Sciencell公司;PCR Mix、DL2 000 DNA Marker、质粒小提试剂盒和DNA凝胶产物纯化试剂盒购自南京诺唯赞生物科技有限公司;RNA提取试剂盒、PerfectStart Uni RT&PCR Kit和5K DNA Marker购自北京全式金生物技术有限公司。
1.3 引物设计
登录NCBI搜索序列号(NC_003210.1)获取Listeriamonocytogenesserotype 4b str. CLIP 80459全基因序列,搜索Gene ID(7703592、7703593和7703594)获取EIIA、EIIB和EIIC基因序列。利用Primer Premier 5设计相关引物(表1)。其中VΔEII-F/R位于EII基因上游725 bp和下游708 bp,用于对EII基因缺失的验证;CΔEII-Phelp-F/R为EII基因的扩增引物;CΔEII-Pnative-F/R为天然启动子与EII基因的扩增引物,其中Pnative为在EII基因上游序列中利用Softberry在线预测的天然启动子。将引物送至生工生物工程(上海)股份有限公司合成。

表1 本研究中所用PCR引物
1.4 EII基因缺失株的构建
根据李红欢[19]方法制备LM928感受态细胞。以LM928基因组为模板,PCR扩增EII基因的上下游同源臂,将电泳凝胶产物纯化后经SOE-PCR获得融合片段,4 ℃过夜连接至pMD19-T(simple)并测序,重组质粒pMD19-T-ΔEII图谱如图1(a),将测序正确的重组质粒pMD19-T-ΔEII与pKSV7在37 ℃经PstⅠ和EcoRⅠ双酶切3 h,4 ℃过夜连接,连接产物转化至大肠杆菌DH5α,涂布于含有氨苄青霉素的LB固体培养基,利用引物ΔEIIup-F和ΔEIIdown-R筛选阳性转化子,再经双酶切鉴定正确的重组质粒pKSV7-ΔEII电击转化入LM928感受态细胞中,重组质粒pKSV7-ΔEII图谱如图1(b),阳性电转子接种于含有氯霉素的BHI液体培养基中,120 r/min,42 ℃振荡培养,每隔5代用引物VΔEII-F/R检测同源重组情况,筛选缺失株。缺失株在无氯霉素和30 ℃的条件下120 r/min震荡培养,以消除pKSV7-ΔEII质粒。每隔5代利用引物VΔEII-F/R进行遗传稳定性的检测,将PCR产物纯化回收后测序,测序正确的缺失株命名为LM928ΔEII。

图1 重组质粒pMD19-T-ΔEII(a)、pKSV7-ΔEII(b)、pIMK2-EII(c)和pIMK2-EII-Pnative(d)质粒图谱
1.5 EII基因回补株的构建
利用引物CΔEII-Phelp-F/R扩增EII基因,与pIMK2经PstⅠ和XhoⅠ双酶切后连接,利用引物CΔEII-Pnative-F/R扩增的EII-Pnative基因,pIMK2经SacⅠ和XhoⅠ双酶切切除了自身的Phelp,再与双酶切的EII-Pnative基因连接,连接产物转化至大肠杆菌DH5α,大肠杆菌转化子用引物EII-F/R与CΔEII-Pnative-F/R验证,得到重组回补质粒pIMK2-EII和pIMK2-EII-Pnative,重组回补质粒pIMK2-EII和pIMK2-EII-Pnative图谱如图1(c)~(d),电转入LM928ΔEII感受态细胞中,经PCR筛选的阳性电转子接种于含有卡那霉素的BHI液体培养基中振荡培养,经PCR与双酶切验证后送测序。测序正确的强启动子回补株命名为CLM928ΔEII-Phelp,天然启动子回补株命名为CLM928ΔEII-Pnative。
1.6 EII基因缺失株及回补株体外生长曲线的测定
将LM928、LM928ΔEII、CLM928ΔEII-Phelp和CLM928ΔEII-Pnative分别划线,37 ℃静置培养后挑取单菌落于BHI液体培养基在37 ℃和160 r/min的条件下振荡培养12~14 h至OD600值为0.8,细菌悬液按照1∶100的比例接种于20 mL的BHI液体培养基,记为0 h,且此时4个菌株OD600数值一致,此后每隔2 h吸取样品至96孔板并设置3个平行,酶标仪读取OD600数值,直至OD600数值趋于稳定。以时间为横坐标,OD600数值为纵坐标,绘制4株菌株的生长曲线。
1.7 EII基因缺失株及回补株在HCMEC/D3细胞内增殖试验
将HCMEC/D3传代至12孔板中,待细胞融合至90%时(细胞数量约为 5×105个/孔),PBS洗涤3次,LM928、LM928ΔEII、CLM928ΔEII-Phelp和CLM928ΔEII-Pnative以感染复数MOI=10感染HCMEC/D3(每组3个平行),感染1 h后更换含有庆大霉素(100 μg/mL)的DMEM培养基以杀死胞外细菌,此时记为0 h,1 h时后更换为含庆大霉素(10 μg/mL)的DMEM培养基继续培养,分别在2、4、6、8、10和12 h使用PBS洗涤,加入胰酶消化,加入0.02%TritonX-100吹打收集样品,10倍梯度倍比稀释后涂布于BHI固体培养基,37 ℃静置培养24 h,菌落计数后分析数据。
1.8 RT-qPCR检测EII的表达以及毒力基因转录水平的比较
1.8.1RT-qPCR检测体外EII基因及关键毒力基因的表达
挑取LM928、LM928ΔEII、CLM928ΔEII-Phelp和CLM928ΔEII-Pnative划线后的单菌落于液体BHI中振荡培养14 h,8 000 r/min,4 ℃离心5 min,收集20 mL菌液,PBS清洗一次,收集的菌液使用液氮研磨,按照细菌总RNA抽提试剂盒进行野生株、缺失株与回补株的总RNA提取,反转录为cDNA,以cDNA为模板,使用PerfectStart Uni RT&PCR Kit检测EII基因及关键毒力基因(prfA、plcA、hly、mpl、actA、plcB、inlA、inlB和inlC)转录水平,收集数据后采用相对定量方法(2-ΔΔCt)分析并使用GraphPad Prism软件进行作图。相关毒力基因以及内参gyrB的基因引物序列见表2。

表2 本研究中所用RT-qPCR引物信息
1.8.2RT-qPCR检测HCMEC/D3细胞内LM关键毒力基因的表达
培养传代细胞至75 cm2培养瓶中,待细胞融合至90%时(细胞数量约为1×107个/瓶),按照1.7的方法感染细胞(每组3个平行),12 h后用PBS收集细胞,再按照1.8.1的方法进行RT-qPCR试验及数据分析。
1.9 数据统计及分析
采用SPSS 26.0统计学软件,组间比较采用单因素方差分析,P<0.05表示差异显著,具有统计学意义;P<0.01表示差异极显著,具有统计学意义。
2 结果与分析
2.1 EII基因缺失株及回补株的构建
经PCR扩增出EII基因上、下游同源臂,大小为526和411 bp,经SOE-PCR融合上、下游同源臂,大小为937 bp(图2(a))。将融合片段ΔEII与pKSV7连接,经双酶切验证,获得937 bp的ΔEII和7 096 bp的pKSV7条带,表明重组质粒pKSV7-ΔEII构建成功(图2(b))。重组质粒电转入LM928感受态细胞,PCR鉴定出大小为937 bp的ΔEII条带(图2(c)),同源重组过程中使用引物VΔEII-F/R检测出大小为1 433 bp的单一短条带,表明缺失株LM928ΔEII构建成功,继续传20代,每隔5代均检测出大小1 433 bp的单一条带,表明LM928ΔEII具有良好的遗传稳定性(图2(d))。重组回补质粒pIMK2-EII和pIMK2-EII-Pnative电转入LM928ΔEII感受态细胞中,使用引物EII-F/R与CΔEII-Pnative-F/R筛选出大小为1 993 bp和2 524 bp的阳性电转子。此结果表明CLM928ΔEII-Phelp与CLM928ΔEII-Pnative构建成功(图2(e))。
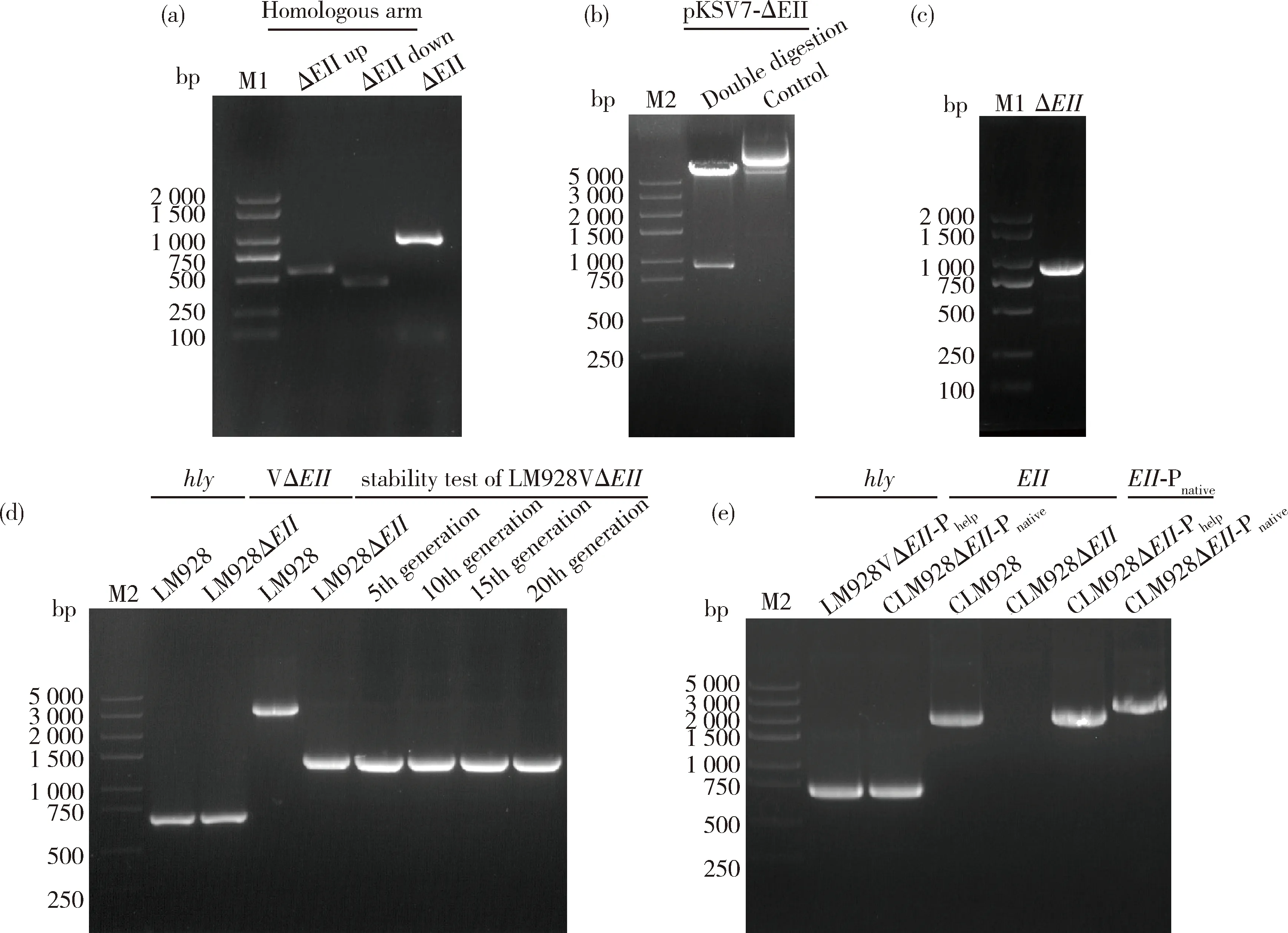
(a)EII上、下游臂同源臂的扩增及同源臂SOE-PCR的扩增;(b)重组质粒pKSV7-ΔEII的双酶切鉴定;(c)重组质粒pKSV7-ΔEII阳性电转子的检测;(d)EII基因缺失株的验证及其遗传稳定性检测;(e)EII基因回补株的鉴定;M1:DL2 000 DNA Marker;M2:Trans 5K DNA Marker。 (a) EII gene upstream and downstream homologous arms and homologous arm SOE-PCR amplification; (b) Identification of recombinant plasmid PKSV7-ΔEII by double digestion; (c) Electroporation positive transformant PCR identification; (d) Verification and stability test of LM928ΔEII; (e) PCR identification of EII complemented strains; M1: DL2 000 DNA Marker; M2: Trans 5K DNA Marker.
2.2 EII基因缺失株及回补株体外生长曲线的测定
在BHI培养基中37 ℃培养的LM928、LM928ΔEII、CLM928ΔEII-Phelp和CLM928ΔEII-Pnative在生长迟缓期、快速生长的对数期和生长平台期生长趋势均无较大差异(图3)。此结果表明EII基因对LM928体外的生长特性无显著影响。
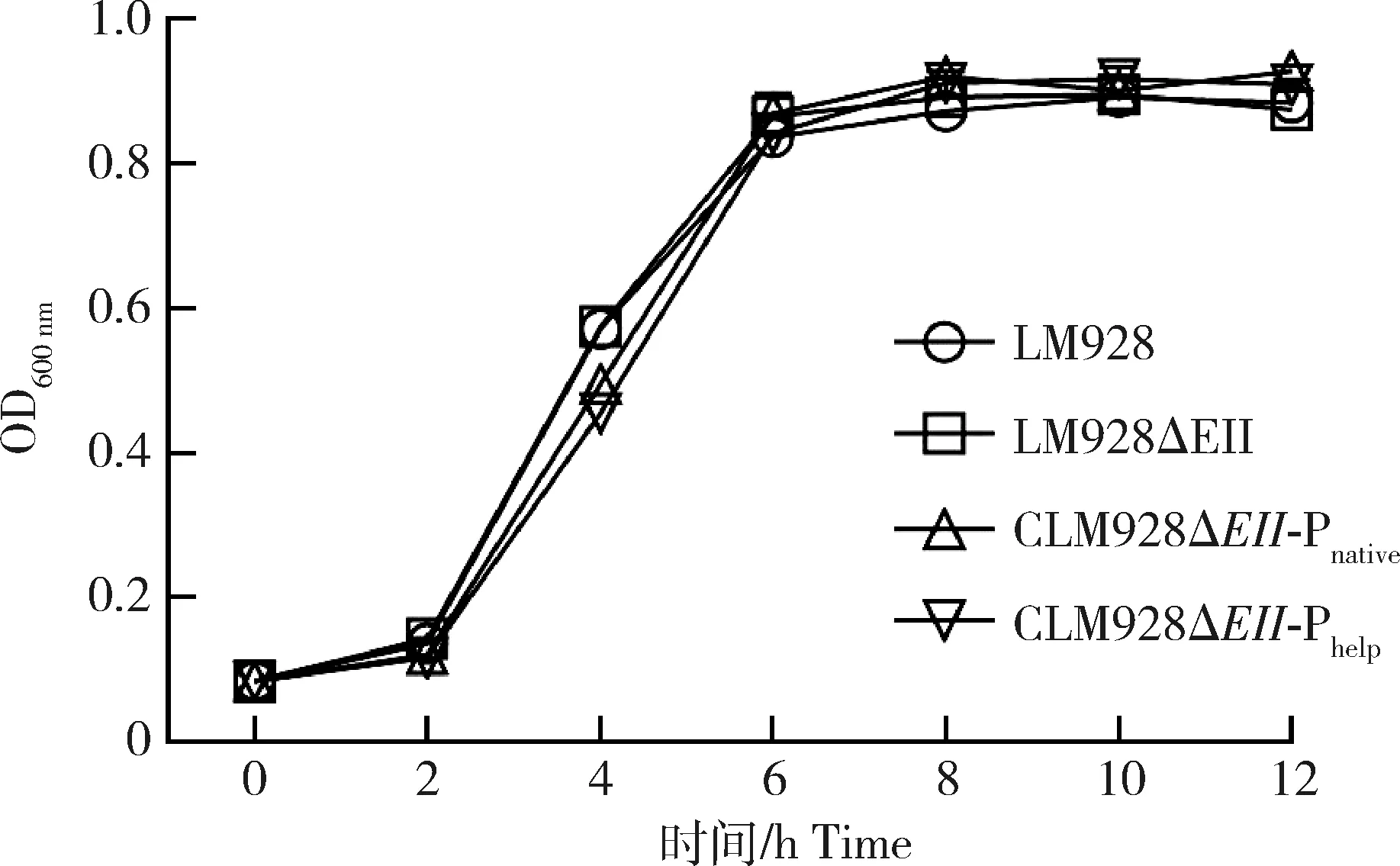
图3 EII基因缺失株及回补株生长曲线测定
2.3 EII基因缺失株及回补株在HCMEC/D3细胞内增殖的测定
使用LM928、LM928ΔEII、CLM928ΔEII-Phelp和CLM928ΔEII-Pnative感染HCMEC/D3,在2、4、6、8、10和12 h收集样品,涂布于BHI固体培养基,37 ℃静置培养24 h,菌落计数。结果显示,LM928ΔEII在8和12 h的细菌载量显著高于LM928(P<0.05),在2和4 h的细菌载量极显著高于LM928(P<0.01);CLM928ΔEII-Phelp在2、4、6、8、10和12 h的细菌载量比LM928极显著降低(P<0.01);CLM928ΔEII-Pnative在2、4和6 h的细菌载量与LM928无显著差异(P>0.05),在8、10和12 h的细菌载量比LM928极显著降低(P<0.01)(图4)。结果表明EII的缺失使LM928在HCMEC/D3中增殖的细菌载量升高,CLM928ΔEII-Phelp在胞内增殖方面未能恢复至LM928水平,而CLM928ΔEII-Pnative在2、4和6 h恢复到了LM928水平。
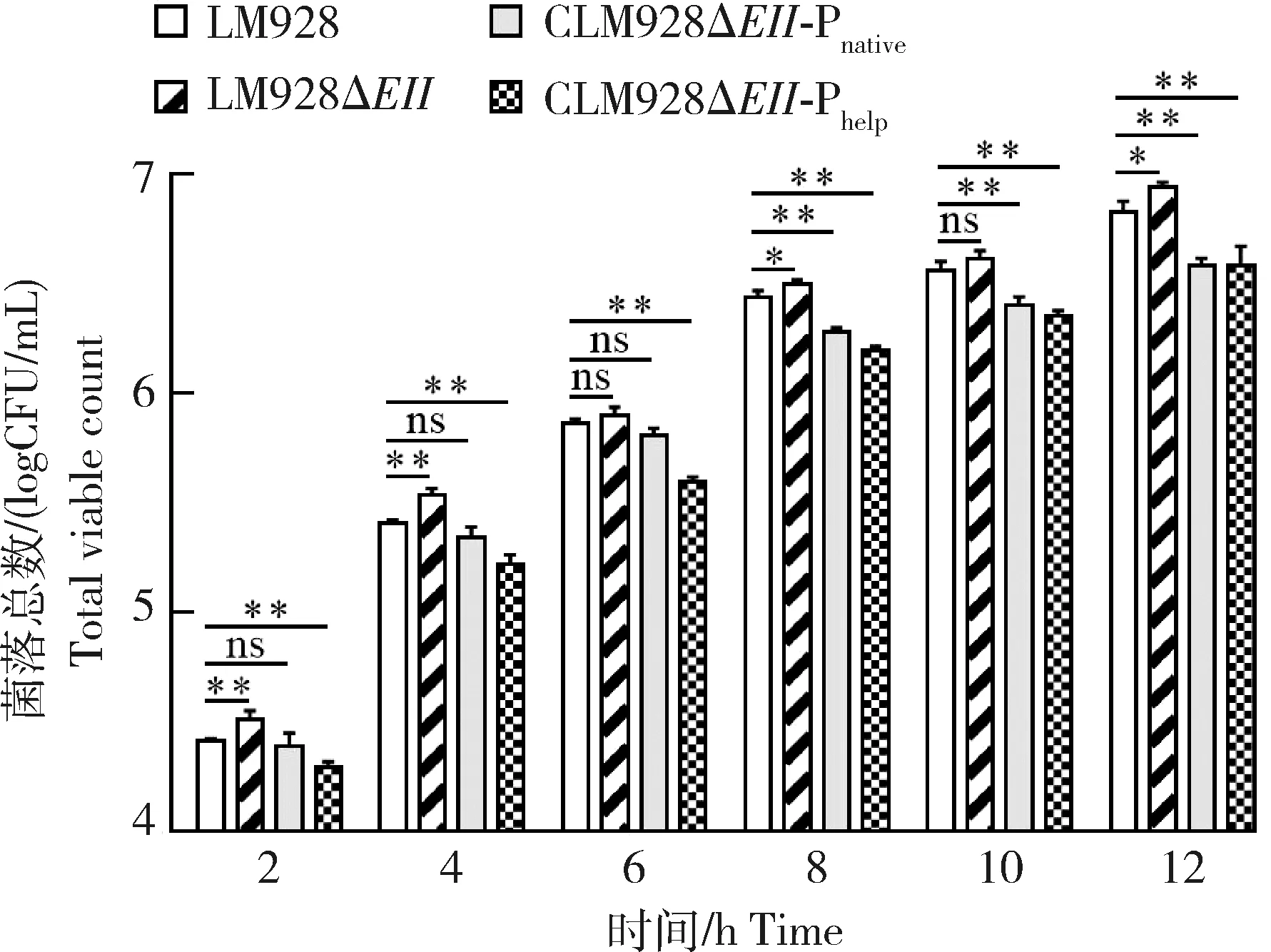
*表示差异显著(P<0.05),**表示差异极显著(P<0.01),ns表示差异不显著。 * means significant difference (P<0.05), ** means extremely significant difference (P<0.01), ns means no significant difference (P>0.05).
2.4 EII对细菌关键毒力基因转录水平的影响
2.4.1RT-qPCR检测细菌EII基因在BHI中的表达提取BHI中振荡培养的LM928、LM928ΔEII、CLM928ΔEII-Phelp和CLM928ΔEII-Pnative的总RNA,采用RT-qPCR方法研究EII基因的表达情况,数据经2-ΔΔCt法分析后作图。结果显示,LM928ΔEII中EII基因相对表达量趋近于0;CLM928ΔEII-Phelp将EII基因极显著表达(P<0.01),其表达量为野生株的9.83倍;CLM928ΔEII-Pnative中EII基因表达与野生株无显著差异(P>0.05)。结果表明LM928ΔEII中EII缺失完全,CLM928ΔEII-Phelp中EII基因过表达,CLM928ΔEII-Pnative中EII恢复至野生株水平(图5(a))。

*表示差异显著(P<0.05),**表示差异极显著(P<0.01),ns表示差异不显著。 * means significant differences (P<0.05), ** means extremely significant differences (P<0.01), ns means no significant differences (P>0.05).
2.4.2RT-qPCR检测细菌在体外培养条件下关键毒力基因的转录水平
提取BHI中振荡培养的LM928、LM928ΔEII、CLM928ΔEII-Phelp和CLM928ΔEII-Pnative的总RNA,采用RT-qPCR方法研究EII基因对关键毒力基因的转录调控,数据经2-ΔΔCt法分析并作图。结果显示,EII基因的缺失使毒力基因plcA、hly、mpl、actA、plcB和inlC极显著上调(P<0.01),prfA、inlA和inlB基因无显著差异(P>0.05)。CLM928ΔEII-Phelp将66.7%(6/9)的基因较野生株表达极显著上升1~4倍(P<0.01),CLM928ΔEII-Pnative使88.9%(8/9)的基因转录水平倍数变化在1倍以内(P<0.01)(图5(b))。结果表明,在BHI中,EII与毒力基因plcA、hly、mpl、actA、plcB和inlC转录水平存在负调控关系,CLM928ΔEII-Phelp与CLM928ΔEII-Pnative均有部分基因表达未恢复至野生株水平。
2.4.3RT-qPCR检测细菌关键毒力基因在HCMEC/D3细胞内的转录水平
使用LM928、LM928ΔEII、CLM928ΔEII-Phelp和CLM928ΔEII-Pnative感染HCMEC/D3,在第12 h收集样品,提取总RNA,采用RT-qPCR方法研究EII基因对关键毒力基因的转录调控。结果显示,EII的缺失使毒力基因prfA、plcA、actA、plcB、inlB、inlC极显著上调(P<0.01),mpl、inlA极显著下调(P<0.01),hly无明显差异(P>0.05)。CLM928ΔEII-Phelp使33.3%(3/9)的基因较LM928极显著上调1~3倍(P<0.01),其中actA上调2.84倍。CLM928ΔEII-Pnative中基因转录水平变化均在1倍以内(图5(c))。结果表明EII与毒力基因prfA、plcA、actA、plcB、inlB和inlC转录水平存在负调控关系,CLM928ΔEII-Phelp有部分基因表达未恢复至野生株水平,CLM928ΔEII-Pnative基因表达基本恢复至野生株水平。
3 讨 论
LM是一种革兰氏阳性土壤细菌,是食源性李斯特菌病的病原体。为了利用腐烂植物和其他生物在土壤中产生的大量碳源,LM拥有大量的碳水化合物转运蛋白[20],其中许多属于PTS。LM共有86个PTS基因,编码29个完整的碳水化合物和糖醇转运PTS,以及多个可能支持化合物转运的单一PTS组分[21]。LIPI-4是一个2016年发现的高毒力基因簇,属于纤维二糖家族PTS[9]。PTS糖特异性转运组分EII除了在糖运输和代谢中的能量互连和信号转导的多功能性,以响应糖的可用性的固有作用外,还与细菌氧化应激、金属离子稳态、耐药性和毒力有关[14-16,22-23]。如甘露糖-PTS的EII(t),在下调LM毒力基因的表达中扮演重要角色[14],在肺炎克雷伯菌中,果糖-PTS中编码EIIC的frwC基因也影响细菌的生成、超黏表型及毒力[23],罗非鱼无乳链球菌纤维二糖-PTS的EIIB蛋白能够负调控弱毒菌株的毒力[24]。有研究发现LM生长在葡萄糖、纤维二糖和果糖等高效代谢碳源上,EII可以抑制PrfA的活性,导致细菌毒力的下降[20,25]。到目前为止,介导纤维二糖和葡萄糖摄取的EII对PrfA活性的抑制最强,而介导甘露糖或果糖摄取的EII对PrfA活性的抑制较弱[25-27]。EII对PrfA活性的抑制作用似乎存在一个层级关系[28],在磷酸化级联反应中,参与碳水化合物运输的EII将磷酸基团转移给输入的碳水化合物,导致去磷酸化的EII水平升高,从而抑制PrfA的活性和LM毒力的表达。
本研究构建了LM928菌株LIPI-4中EII基因的缺失株、强启动子回补株以及天然启动子回补株。生长曲线表明EII的缺失与过表达对LM928在BHI中的生长曲线无影响。但在BHI培养条件下,EII的缺失使依赖PrfA转录的hly、plcA、plcB、actA和mpl的转录水平表现出极显著的上调,但介导LM毒力基因表达的主调控因子prfA的转录水平无显著差异。Stoll等[21]发现PrfA在BHI培养的LM中检测出较低的活性,而在哺乳动物宿主细胞内培养的LM中检测出较高的活性,因此本研究通过感染HCMEC/D3得到了LM在细胞水平上毒力基因的转录水平,缺失EII后,毒力基因prfA、plcA、plcB、inlB、inlC和actA的转录水平极显著上调,与Stoll一致,其中actA的转录水平严格依赖于PrfA的调控[29],prfA上调了0.20倍,actA却上调了2.34倍,这与Joseph等[28]报道的prfA转录水平的小幅升高能够导致依赖于PrfA转录的毒力基因转录水平的大幅增加结果一致。由PrfA调控的基因其蛋白表达能力的大小随着PrfA调控子的浓度和其与启动子亲和性的不同而不同,对于hly、mpl和inlAB,尽管它们都受到PrfA的调控,但是由于启动子对RNA聚合酶的亲和力以及5′非翻译区结构的不同可能会引起这些依赖于PrfA的基因表达水平不同[6]。这可能是本研究中毒力基因hly、mpl和inlA的转录水平未表现出随着prfA表达的升高而升高的原因。在胞内增殖试验中,缺失EII使LM在HCMEC/D3中的细菌数量升高,这与EII缺失使LM内化进入非吞噬细胞系的关键基因(inlB),参与吞噬体液泡逃逸的基因(plcA和plcB)和参与细胞间传播的基因(actA和inlC)转录水平的上调结果相一致。LIPI-4的EII抑制prfA的表达,调控关键毒力因子的转录水平,这与介导纤维二糖和葡萄糖摄取的EII抑制PrfA活性结果[25]一致。EII的缺失使LM关键毒力基因表达上升,这促进了LM进入细胞以及在细胞内的增殖,但对于LM在体外的生长影响无显著差异,这导致了LM在体外与细胞内的增殖情况有所差异。
Monk等[30]构建了整合型LM载体pIMK2,其自身携带的强启动子会提高基因表达的总体水平,pIMK2现已被广泛用于LM回补株的构建。但本研究结果表明CLM928ΔEII-Phelp与LM928的水平差异较大,可能是强启动子使EII的过量表达造成了试验结果的不稳定,表明利用载体提供的强启动子构建回补株可能达不到对缺失株功能补充验证的目的。有研究构建了LM毒力基因的天然启动子回补株,在生化特性与细胞试验中恢复到了野生株的水平[31]。于是本研究选择构建天然启动子回补株,使用生物信息网站Softberry预测EII基因上游非编码区的自身启动子,根据启动子与转录因子结合的顺式元件基序的打分情况,选择了转录起始位点到其上游531 bp的区域作为天然启动子区域,其中包含了两个预测到的启动子,一是位于转录起始位点上游35 bp的核心启动子,包含了两个能决定开启下游基因转录的RNA聚合酶sigma因子RpoD17(结合位点为ATTTTGTA和TTTTGTAT)和RpoD18(结合位点为AATTGAGG)的结合位点,还有一个介导细菌抵抗氧化应激作用的转录调控因子OxyR(结合位点为GATTAATT)和一个几丁质酶与二糖酶的转录调控因子Nagc(结合位点为GAAATAAG)的结合位点;二是位于转录起始位点上游482 bp的近端启动子,包含硝酸盐应答转录调节因子NarL(结合位点为TGCTCCTT)的结合位点。
RT-qPCR结果显示,强启动子将EII过表达,其转录水平上升9.83倍,提示其可作为过表达株研究EII基因,而天然启动子将EII的表达恢复至野生株水平。在缺失EII后,毒力基因表达显著上调,而CLM928ΔEII-Pnative则可以将这些基因恢复或部分恢复,说明EII是LIPI-4中重要的毒力相关基因;有意思的是,当EII基因过表达后,与野生株相比,除了inlA以外,其他毒力基因也全部上调,这一方面进一步确定了EII基因参与LM的毒力调控。另一方面,EII可能具有直接平衡或控制毒力的能力:EII表达量过低(缺失条件下),解除对PrfA的抑制,直接调控毒力因子转录水平的上升;但当EII表达量过高时(过表达),降低了透性酶在膜上的定位,且EII过量表达后,产生复杂的产物,导致EII基因甲基化,EII的活性被抑制,从而解除了EII对PrfA活性的抑制,导致毒力基因表达的升高,这也可能是过表达回补株与缺失株毒力基因转录水平相近的原因。在体外和细胞内的培养下,CLM928ΔEII-Pnative的毒力基因转录水平基本恢复至野生株水平,这与天然启动子将EII的表达恢复至野生株水平一致,而actA等基因的转录水平没有恢复至野生株水平可能是因为本研究构建的天然启动子只选择了核心启动子和近端启动子,没有包含的远端启动子也可能影响了毒力因子的表达。胞内增殖试验中,CLM928ΔEII-Phelp和CLM928ΔEII-Pnative都表现出了增殖的下降,可能是由于外源质粒的插入,LM减少增殖来维持质粒的复制所导致的。以上提示,CLM928ΔEII-Pnative更适合作为阳性对照株用于对LM928缺失株功能的补充验证。
本研究只局限于体外试验,后续不仅要研究EII对HCMEC/D3细胞通透性的影响,还需要以小鼠为模型进行感染试验,通过体内实验对EII与LM之间的致病机制进行研究。
4 结 论
本研究构建EII缺失株、强启动子回补株和天然启动子回补株,通过胞内增殖实验和检测体外与细胞内的毒力因子转录水平以深入了解EII对LM毒力基因表达的影响,结果表明LIPI-4EII对LM关键毒力因子的转录具有负调控作用。本研究为进一步探究LM LIPI-4致病机理提供相关的理论依据。
