非水饱和相矿物表面催化转化有机污染物的研究进展
2023-06-25黄姝晗范振辉谷成金鑫
黄姝晗,范振辉,谷成,金鑫
(南京大学环境学院,南京,210023)
以层状硅酸盐矿物和各类含铁、铝氧化物为代表的天然矿物广泛分布于土壤、大气、水体等环境中[1-5].天然矿物能够有效地吸附、转化污染物,成为影响环境中有机污染物迁移转化的重要媒介[6-10].在自然条件下,土壤或大气矿物颗粒表面时常处于水分非饱和状态[11-13].在干旱条件下,土壤微生物活性会受到极大的抑制[14],而由土壤矿物诱导的污染物非生物转化作用反而增强.因此,其环境意义得到凸显.研究非水饱和相条件下天然矿物对污染物的催化转化效应更能反映实际土壤或大气中污染物的迁移转化过程.此外,不同的矿物表面在非水饱和相条件下会表现出不同表面性质,因此对污染物的催化作用机制不尽相同[15].阐明非水饱和相矿物表面催化机制有助于进一步理解污染物的环境行为,也有助于发展有效的环境修复手段.
1 典型天然矿物
矿物按成分主要分为硅酸盐矿物、氧化物矿物、硫化矿物、硼酸盐矿物和其他矿物[9].
1.1 层状硅酸盐矿物层状硅酸盐矿物是指以硅氧四面体和铝氧八面体为基本单元组成的黏土矿物,其粒径一般小于1 μm,除海泡石、坡缕石外,一般均具层状结构[16].硅氧四面体或铝氧八面体聚合后在二维方向上延伸排列形成硅氧片层和铝氧片层.单位晶片纵向叠加形成晶层,按照晶层组成可以把层状硅酸盐矿物分为1∶1 型和2∶1型.前者由一层铝氧层叠加一层硅氧层组成,后者由一层铝氧层和两层硅氧层组成三明治结构.层状结构使黏土矿物拥有两种表面类型——基底表面(层间表面)和边缘表面[17].基底表面具有固定的负电荷,电荷量由黏土结构类型决定;边面的金属氧化物或硅氧化物断层带有羟基,其电离程度取决于表面溶液相的pH[18].
1.1.1 1∶1 型黏土矿物由硅氧片和铝氧片堆叠组成,晶层与晶层间连接紧密,无膨胀性,电荷量少,胶体特性弱,包括高岭石、珍珠陶土、迪恺石及埃洛石等.高岭石(Kaolinite)(图1)是地球上存在最广泛的硅酸盐矿物之一,化学式为Al4Si4O1(0OH)8.有限的电荷量和较小的层间距导致其吸附和离子交换能力较差[19].其最活跃的基团是表面羟基(-OH),它可以参与广泛的化学过程,相较于硅氧面,铝氧面上羟基更多且活性更高[9,20-21].此外,高岭石晶格边缘有许多破碎的化学键,容易吸附负电荷、产生更高活性的边缘羟基,这也被认为是高岭石表面酸性来源之一[22-23].埃洛石(Halloysite)是一种天然高岭土水合物,化学式为Si4Al4O1(0OH)8·4H2O.它是一种管状矿物,内层为铝氧层,外层为硅氧层,内外层具有不同的化学性质[24-26].埃洛石在70~100 ℃下可脱水,脱水后形态与高岭石类似[27].

图1 高岭石的结构示意图[20]Fig.1 Representation of the Kaolinite structure (after ref.[20])
1.1.2 2∶1 型黏土矿物其结构由两层硅氧片夹一层水铝片构成,片层间空间较大,可容纳水分子或阳离子,包括蒙脱石、伊利石、绿脱石、拜来石、蛭石等.按照其膨胀性,又可分为膨胀性矿物和非膨胀性矿物.2∶1 型膨胀性矿物以蒙脱石(Montmorillonite)为主(图2),同晶置换作用导致其带有永久负电荷,层间存在可交换的水合阳离子,可以发生水合作用从而具有膨胀性.蒙脱石化学式为Al4Si8O2(0OH)4·nH2O,由于其高阳离子交换容量(Cation Exchange Capacity,CEC)、大比表面积和膨胀层间结构,蒙脱石黏土对污染物有较强的吸附和催化转化能力[28-30].其表面性质因水分含量和可交换阳离子不同而存在差异,当蒙脱石表面含水量较低时,可交换阳离子会对配位的水分子产生极化作用,使表面布朗斯特(Brönsted)酸性增强[18,31-32],离子水合程度降低也有利于促进表面路易斯(Lewis)酸性位点与污染物发生作用[15].非膨胀性矿物以伊利石为主,化学式为K(2Al·Fe·Mg)(4SiAl)8O2(0OH)4·nH2O.在伊利石晶层之间吸附有钾离子,对相邻两晶层产生了很强的键联效果,使晶层不易膨胀,同时,伊利石也带有大量电荷.此外,绿泥石是一种2∶1∶1 型硅酸盐黏土矿物,其结构与蒙脱石类似,但富含镁、铁及少量铬,同晶置换较为普遍,元素组成的变化较大.
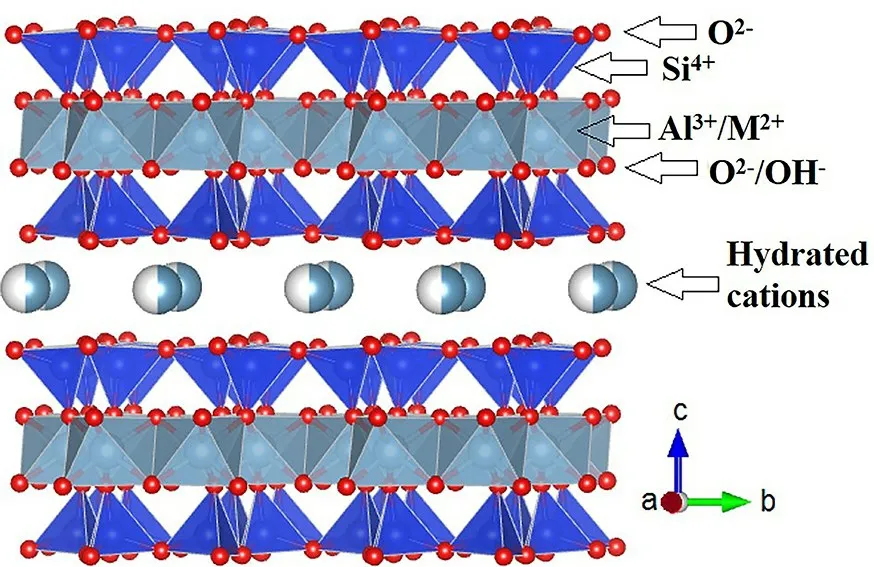
图2 蒙脱石结构示意图[20]Fig.2 Representation of the Montmorillonite structure(after ref.[20])
1.2 铁、铝氧化物铝、铁是地壳中除氧、硅外含量最高的元素.除了构成硅酸盐黏土矿物的基本结构以外,环境中铝元素也以多种氧化物或水合氧化物的形式存在.土壤中氧化铝的形态主要为α‐氧化铝(α‐Al2O3)和γ‐氧化铝(γ‐Al2O3).α‐Al2O3俗称刚玉,结构紧密,性质稳定.γ‐Al2O3孔隙度高,表面积大,表面存在多种羟基[33].氧化铝在水分作用下表面会迅速羟基化,改变其表面结构,其反应活性也受到表面羟基化的强烈影响[34-35].
土壤中的铁元素主要以氧化物和氢氧化物形式存在,也有少量碳酸盐、硫化物和硫酸盐含铁矿物[36].土壤中最常见的氧化铁矿物是赤铁矿(α‐Fe2O3)和针铁矿(α‐FeOOH),主要呈胶膜质包被在土壤颗粒的表面[37].赤铁矿是最稳定的氧化铁矿物,表面羟基较少.针铁矿呈针状,表面有大量的羟基,可有效促进有机污染物转化[38].菱铁矿(FeCO3)则是一种较稳定的亚铁矿物.此外,非晶质(无定形)的铁氧化物(如水铁矿)是土壤和沉积物中吸附有机物和阴离子的主要贡献者[39-41].铁、铝氧化物晶型和晶面组成与其反应活性直接相关,不同晶型甚至同一晶型不同晶面结构都会导致反应的显著差异[42-44].
2 矿物表面酸碱特性及其影响因素
土壤矿物表面的酸碱特性及配位作用已被多项研究证明是诱导有机污染物发生催化转化的关键作用机理,包括Brönsted 酸、Lewis 酸作用和表面羟基形成的氢键作用等.
2.1 Brönsted 酸性及其影响因素Brönsted 酸性指矿物表面给质子的能力,来源于矿物边缘羟基(≡AlOH,≡Fe(OH)n,≡SiOH)和基面配位氧原子羟基的质子解离[18],其强度取决于暴露的金属离子的极化能力、配位数、表面含水量[45].蒙脱石层间可交换阳离子可以极化配位的水分子发生质子解离,从而产生极强的Brönsted 酸性[18,46-47].由层间阳离子诱导的表面Brönsted 酸性随着层间阳离子的电荷与半径比的增加而增加(Al3+>Mg2+>Ca2+>Li+>Na+>K+),随着层间含水量的降低而增加.当黏土矿物表面仅覆盖单层水分子时,表面极化程度达到最高[18,45,48-50].该现象不仅体现在黏土矿物上,在TiO2和氧化铝上也存在类似现象[51-52].此外,黏土矿物表面暴露的Si4+以及同晶替换作用产生的晶格Al3+,Fe3+等在水合时可以产生弱的Brönsted 酸性位点[53].相较于Si-OH-Si 和Al-OH-Al,Si-OH-Al 产生的Brönsted 酸性更强.干燥脱水可以极大地增强其酸性,甚至使这些Brönsted 酸位点转化为Lew‐is 酸位点[53].
矿物结构也对Brönsted 酸性产生较大影响.Wei et al[54]的研究表明,表面酸度受电荷位置影响,硅氧四面体同晶置换产生的负电荷比八面体中的更接近层间空间,因此相较于伊利石、蛭石等,蒙脱石的表面酸性更强、催化水解能力更显著.
2.2 Lewis 酸性及其影响因素黏土矿物Lewis酸性位点来源于晶格结构中金属阳离子(如暴露在晶体边缘的Al3+)或层间可交换的过渡金属离子(如Fe3+,Cu2+)[30,47,49,55-56].对于铁、铝氧化物而言,其表面配位不饱和晶格Fe3+和Al3+具有一定的Lewis 酸性.表面O2-带来的表面羟基既可以产生氢键作用,又可以提供质子[57].Lewis 酸作用能够诱导配位的有机物发生电子结构重排,降低反应活化能[58].在Lewis 酸与某些有机污染物作用时,可以观察到阳离子自由基的产生,并进一步介导连续聚合反应[59].Lewis 酸性的强弱取决于离子的水合程度[60-61].在极度干旱条件下,相较于Brönsted酸,Lewis 酸对反应的催化作用更为显著[62].
黏土矿物表面同时具有Brönsted 酸性和Lei‐ws 酸性位点,极高的表面酸度还可诱导有机污染物发生酸性水解.Xu[63]报道了在Lewis 酸催化高岭石对聚(二甲基硅氧烷)的高效降解中,水解速率取决于矿物表面酸性位点.Wei et al[54]研究了多种矿物对氨基甲酸酯类农药的水解,得出表面Brönsted 酸性位点和表面螯合物的形成都是矿物催化水解的关键因素.此外,黏土矿物表面酸性位点还可以催化污染物氧化裂解、光解[59,64].
铁、铝氧化物以Lewis 酸为主[65].Mäkie et al[66-67]的研究表明,赤铁矿和磁铁矿上的Lewis 酸性位点可以络合磷酸酯,针铁矿上的Lewis 酸性位点能提供比羟基形成的氢键作用更强的吸附能力.γ‐Al2O3上也有类似的研究结果[68].Torrents and Stone[69]证明,过渡金属(如Fe 和Ti)上Lewis酸性位点在催化水解羧酸酯中的作用.Wu et al[70]的研究也证明了在干燥条件下赤铁矿、磁铁矿对氯霉素的催化水解能力主要依靠Lewis 酸催化,而针铁矿同时存在表面羟基的氢键作用和Lewis 酸作用两种机理.
2.3 湿度对矿物表面酸碱性的影响表面湿度对于矿物表面酸碱性有着显著影响,总的来说,黏土矿物表面酸度随着湿度升高迅速下降[63,71].
Brönsted 酸在水相条件下根据环境pH 仅表现出中等强度的酸性,而在非水饱和相条件下(含水量小于5 wt%)矿物表面形成的酸度接近浓度为90%的浓硫酸[15].这是因为矿物表面水分子越少,阳离子对剩余水分子产生的极化作用越强,矿物表面酸性越强[32,51,72-73].而在水相条件下,矿物表面和层间的多层水分子的氢键网络会消除这种极化现象.在这种情况下,表面酸度主要受边缘羟基的pKa控制(pH=4~8),因此水溶液中黏土矿物表现为中性至弱酸性[74-75].
Liu et al[45],Voudrias and Reinhard[56],Solo‐man et al[76]在1971 年借助红外光谱表明,当矿物表面仅含1 wt%的游离水时只展现Brönsted 酸性,而随着进一步干燥脱水,矿物表面会产生更强的Lewis 酸性,并增强邻近Brönsted 酸性位点.在极度干燥下(表面自由水甚至表面羟基脱除),Lewis 酸性会大大增强,相反Brönsted 酸性则减弱[50,77-79].Kikuchi et al[77]在研究中发现,高温处理会使矿物的酸性位点减少,并促使Brönsted 酸性位点转化成Lewis 酸性位点,成为反应的主要活性位点.同时层间游离水会和有机分子争夺层中的Lewis 酸性位点并影响有机分子的电离势,从而影响由Lewis 酸催化的矿物表面反应[15,56].
3 非水饱和相矿物表面反应
天然矿物表面性质复杂多样,已有许多研究关注并证明了矿物界面上发生的多种环境过程,包括吸附、螯合、光解、水解、氧化还原等[30,80-83].矿物在非水饱和相条件下会展现出不同的表面特性,湿度对其具有强烈影响,因此污染物在非水饱和相矿物表面反应可能与在水溶液中明显不同.类似的观点最早在1970 年由Mortland 提出,他认为自然环境中可能同时存在黏土矿物在水溶液中和在干燥条件下与有机物的相互作用[84].同时,土壤实际上往往处于非饱和状态(含水率为2 wt%~60 wt%)[12],因此,研究非水饱和相中的反应过程更能解释自然状态下土壤中污染物的环境行为.
3.1 非水饱和相矿物表面水解反应
3.1.1 水解不同有机农药20 世纪70 年代Saltzman et al[85-86]和Mingelgrin et al[87-88]率先开展了非水饱和相条件下矿物表面催化水解污染物的研究.他们以有机磷农药为研究对象,探讨了湿度对以高岭石为主的黏土矿物表面催化水解能力的影响,指出高岭石催化水解有机磷农药的最佳含水率范围是2 wt%~10 wt%,而在较高湿度条件下或水溶液中其水解能力会受到一定程度的抑制.蒙脱土的催化水解能力主要来自层间可交换阳离子,阳离子类型与矿物表面水分含量极大地影响水解速率.El‐Amamy and Mill[89]研究了不同含水率下蒙脱石和高岭石表面酸性对环氧丙烷等农药的催化水解(图3),结果表明高岭石和蒙脱石的催化水解存在不同最佳湿度区间(高岭石:2 wt%~10 wt%,蒙脱石:10 wt%~50 wt%),且腐殖酸对高岭石的催化水解反应有明显抑制作用,而蒙脱石则不易受到干扰.他们指出这种矿物表面效应并不局限于特定化合物,在所有被H3O+或HO-催化的可水解结构上都可能发生这种现象.上述研究在一定程度上揭示了非水饱和相条件下矿物对有机农药的水解过程,但对机理的阐释不足,有待新的实验手段进一步证明.
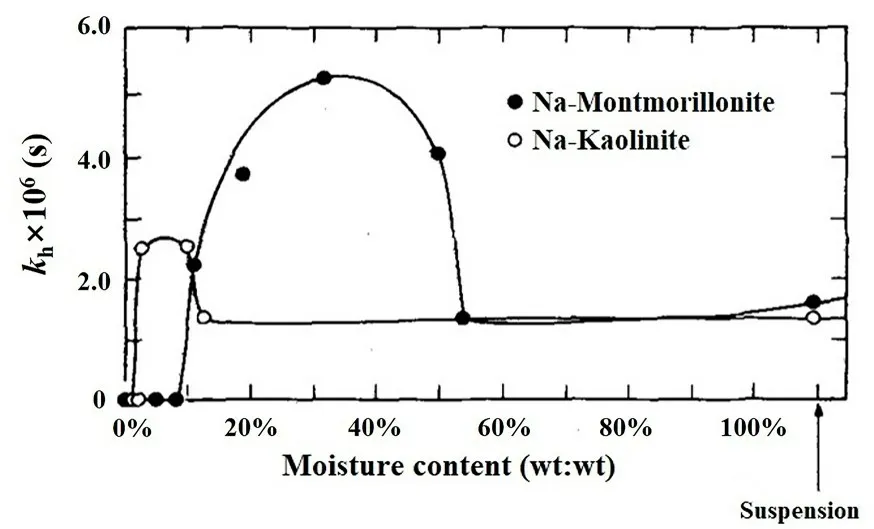
图3 环氧丙烷在钠饱和蒙脱石和高岭土上的水解速率[89]Fig.3 Rate of hydrolysis of epoxypropane on Montmo⁃rillonite and Kaolinite saturated with various cations (af⁃ter ref.[89])
2011 年Mäkie et al[66-67]利用原位漫反射红外傅里叶变换光谱(DRIFT)研究了有机磷农药在三种铁矿上的吸附,发现在赤铁矿和磁铁矿上的吸附机制主要依赖于Lewis 酸性位点,而在针铁矿上则出现了由Brönsted 酸性位点介导的氢键作用.同时,在经过低湿处理(相对湿度,Relative Humidity,RH=17%)后,干燥针铁矿表面观察到了磷酸钠三甲酯的缓慢水解.
3.1.2 水解聚(二甲基硅氧烷)1979 年Buch and Ingebrigtson[90]提出在干燥的矿物或土壤表面聚(二甲基硅氧烷)(PDMS)会经历广泛的水解过程.1994 年起Lehmann et al[91-92]采取了一系列土壤实验证实了这一现象,他们选取了来自不同地区的七种不同土壤来判断湿度对PDMS 水解的影响,发现PDMS 的水解对湿度敏感.水解过程在潮湿土壤中缓慢进行,随着土壤从潮湿逐渐到干燥,PDMS 迅速水解并最终被降解为水溶性单体——二甲基硅烷二醇(DMSD).若重新润湿干燥土壤,水解反应又会被重新抑制[93].且在田间实验中,PDMS 在夏季干燥土壤表面的降解速率要比秋冬大得多[94-95].在Lehmann et al[91-92]实验中,尽管作者推测反应机制可能是非生物的,但其并未对土壤进行灭菌处理,也并未了解土壤的催化机制.因此,Xu and Lehmann[96]在32%的相对湿度(RH)下用同位素标记的方法开展了14C‐PD‐MS 在12 种黏土矿物上的降解实验,并进一步探讨了可交换性阳离子和湿度对降解速率的影响[63].结果表明不同的黏土矿物对PDMS 有着不同的催化活性,同一矿物上降解速率随着可交换阳离子(即Al3+,Ca2+,Na+)极化能力的增加和湿度的降低而增加,据此他们提出表面Brönsted 酸性位点参与催化水解PDMS.而高岭石等低阳离子交换量的黏土矿物上的PDMS 高水解速率则被解释为Lewis 酸性位点的作用,作者推测晶体边缘的铝醇基或结构铝可能是关键催化位点.
3.1.3 水解抗生素和邻苯二甲酸酯学者们也系统地研究了非水饱和相条件下新型有机污染物在高岭石、蒙脱石、铁氧化物等矿物表面的转化过程,并通过光谱分析和计算化学等手段对其机理进行了详细地阐释[58,70,97-98].他们指出非水饱和相下矿物表面催化水解存在多种催化机制,包括Brönsted 酸、Lewis 酸作用和表面羟基形成的氢键作用等.研究结果表明(图4a),高岭石铝氧面羟基在中等相对湿度(RH为33%~76%)下可以与氯霉素(Chloramphenicol,CAP)的酰胺键作用产生氢键,从而显著催化氯霉素水解[58].该结果与El‐Amamy and Mill 的研究结果一致[89],即矿物表面有限的水分子可以协助氯霉素扩散到黏土表面的反应位点,而过量的水分子则会竞争反应位点.对于阳离子交换的蒙脱石而言(图4b~d),Fe3+或Al3+交换的蒙脱石展现出催化活性最强,且具有两种催化机制,Brönsted 酸机制在低含水率(RH<10%)下主导催化反应,Lewis 酸机制则在较高含水率(RH为10%~100%)时占主导.在铁氧化物上也能观察到类似的现象,即在RH为33%~76%时,磁铁矿等四种铁氧矿物都能显著催化氯霉素的水解,由于铁矿表面特性不同,其催化过程也有明显差异(图5)[70].磁铁矿上表现出最强的Lewis 酸性,因此具有最强的催化活性.针铁矿的催化能力主要依赖于表面羟基与氯霉素形成的较弱的氢键作用.而在菱铁矿上则通过形成双齿氢键螯合作用诱导氯霉素的水解.

图4 不同相对湿度下CAP 在(a)钙饱和高岭土(Ca⁃KGa2)和(b)钙饱和蒙脱石(Ca⁃MMT)上的降解动力学(25 ℃)[58];(c)在RH 为76%时,CAP 在不同阳离子交换蒙脱石上(Mn+⁃Mt)的水解动力学(55 ℃)[97];(d)不同阳离子交换蒙脱石上(Mn+⁃Mt)CAP 的水解速率常数(25 ℃)与黏土含水量的关系[97]Fig.4 Degradation kinetics of CAP on (a) Ca⁃KGa2 and (b) Ca⁃SWy2 under different RHs at 25 °C (after ref.[58]),(c) the hydrolysis kinetics of CAP on different cation⁃exchanged montmorillonites (Mn+⁃Mt) at RH=76% and reaction temperature of 55 ℃ (after ref.[97]),(d) The hydrolysis rate constants of CAP (at 25 ℃) as a function of clay water contents for different cation⁃exchanged montmorillonites (Mn+⁃Mt) (after ref.[97])
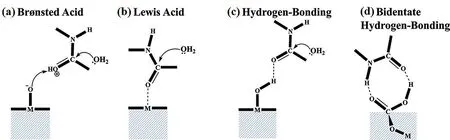
图5 铁氧矿物表面与CAP 的不同结合位点[70]Fig.5 Different interaction sites between iron mineral surfaces and CAP (after ref.[70])
除了氯霉素在干燥的矿物表面发生的水解反应外,Jin et al[98]继续研究了低湿度下晶面效应对赤铁矿水解邻苯二甲酸酯的影响,发现在非水饱和相条件下污染物的水解明显快于水相条件,且晶面上Fe 位点的不同排布会产生不同的Lewis酸性,从而使催化活性产生较大差异.研究为具有更高活性的催化材料的研发提供了新的机制.
3.2 非水饱和相矿物表面氧化聚合反应有机分子可以吸附在层状黏土矿物的中间层中,并在层间表面与过渡态金属阳离子相互作用产生有机阳离子自由基,这些阳离子随后可与自身相互作用形成更高分子量的二聚体或多聚体,在矿物表面脱水后聚合现象会更加明显[99-100].这种有机污染物在黏土矿物表面发生的氧化聚合反应可能是自然界中生成腐殖质或者大气环境中褐碳来源的重要途径,同时也是二噁英类物质潜在的天然形成途径[101-104].
3.2.1 与芳香族化合物反应生成二聚体或多聚体物质自Doner and Mortland[105]首次报道了Cu2+‐蒙脱石干燥表面吸附产生苯络合物以来,大量研究证明了矿物表面有色芳烃络合物的形成[106-110].电子自旋共振谱和红外光谱表明芳香族分子和Cu2+‐蒙脱石可形成两种不同的层间络合物:Ⅰ型络合物苯环完整,具有芳香性;Ⅱ型络合物(阳离子自由基)苯环扭曲,芳香性受到干扰[56,106].形成Ⅱ型络合物时的矿物水合程度低于Ⅰ型,因此可以通过控制矿物水合程度促使两种络合物相互转化[106,109,111-113].Soma et al[114-115]对反应中水的作用机理进行了阐释:水可以与有机分子竞争层间吸附位点或与阳离子自由基反应使其恢复至中性分子形态,但水的作用是可逆的,在重新失水条件下反应可以继续进行.拉曼光谱和电子自旋共振谱表明在阳离子交换蒙脱石表面吸附形成络合物的过程中伴随着层间金属阳离子的还原,聚合反应强度与交换性阳离子氧化电位成正比(Fe3+>Al3+>Ca2+>Na+)[113-114,116-118].
此外,非水饱和相黏土矿物表面芳香族物质的分子结构也能影响形成的络合物类型.苯环上取代基不同,形成的自由基种类不同.当苯对位被取代基占据时仅形成母体分子的阳离子自由基,而其他单取代苯可在对位二聚形成联苯型阳离子,并可进一步生成聚对苯基阳离子[113-115,118-119].如果反应物是卤代酚类物质,则有可能生成剧毒的二噁英.
Boyd et al[104,120]观察到在Cu2+‐蒙脱石上吸附五氯酚(PCP)和二噁英时阳离子自由基的形成,质谱显示,这些阳离子自由基是母体分子的二聚体和三聚体.作者认为这些低聚反应可能成为降解或转化有机氯代污染物生成二噁英的关键步骤.Gu et al[101]在2008 年发现Fe3+‐蒙脱石可在环境条件下催化五氯酚(PCP)自发生成八氯二苯并对二噁英(OCDD),首次揭示了黏土矿物原位催化形成类二噁英类物质的过程.实验证明反应在较低相对湿度下(RH为5%~18%)才能发生,且OCDD 的形成与系统的湿度呈反比.随后Gu et al[102,121]采用傅立叶光谱(FTIR)和量子力学等方法确定了反应机理(图6)并且原位还原了水分子对反应的影响,黏土矿物中间层中的可交换Fe3+可充当氧化剂触发氯酚类物质的单电子转移,生成五氯酚阳离子自由基,从而引发后续聚合反应.傅立叶红外吸收光谱结果与Soma et al[114-115]的研究结果一致:水会与有机分子竞争金属阳离子的配位点,因此阳离子自由基的形成受湿度的影响极大.Gu et al[101,121]进一步研究了黏土矿物结构铁和层间可交换铁的反应差异,对于八面体结构铁,体系湿度有利于反应的进行.这是由于高湿条件下(RH为54%~75%)水分子可与反应中间体形成氢键降低反应活化能.在Wang et al[122]开展的结构铁诱导2,4,6‐三氯苯酚(2,4,6‐TCP)生成羟基化多氯联苯醚(HO‐PCDEs)的反应中,除了反应湿度外,结构铁在黏土晶格中的位置和畸变程度亦会对反应产生显著影响.此外,Wang et al[122]还证明通过卤代酚在黏土矿物上氧化聚合形成的二噁英类化合物的毒性和产量在很大程度上取决于携带的卤素原子的数量、种类和位置,其中卤素原子的位置和种类对催化反应的影响比取代基数量的影响更显著,邻位取代通常比间位取代反应活性更高.溴酚比氯酚反应活性更强,也更容易受到湿度的影响[123].

图6 在Fe(Ⅲ)改性/饱和蒙脱石上生成二噁英/类二噁英化合物和EPFR 的反应机理示意图[127]Fig.6 Schematic of the reaction mechanism for the formation of dioxin/dioxin⁃like compounds and EPFRs on Fe(Ⅲ)⁃modified/saturated montmorillonite (after ref.[127])
在2010 年Liyanapatirana et al[124]就已发现在RH为40%的条件下阳离子交换的蒙脱石可催化三氯生转化产生二聚体或三聚体,且与Na+‐蒙脱石相比,Fe3+‐蒙脱石的催化聚合能力更强.随后Ding et al[125]的研究证明在非水饱和相条件下铁、锰氧化物可催化三氯生进一步聚合生成二噁英类化合物.在该反应中铁、锰氧化物首先通过电子转移介导三氯生产生自由基中间体(如图7),较低湿度条件(RH<8%)能促进产生的中间体通过分子内亲核取代反应生成2,8‐二氯二苯并对二噁英(2,8‐DCDD),而较高含水率(50 wt%)则有利于自由基醚键的断裂[125].非水饱和相条件下湿度对铁、锰氧化物催化羟基化多溴联苯醚(HOPBDEs)发生氧化聚合生成1,3,8‐三溴二苯并对二噁英有类似影响[38,125].此外,软锰矿也已被证实能够在非水饱和相条件下(RH为2%~90%)介导2,4,6‐三氯苯酚氧化聚合,这也是环境中形成二噁英类化合物的另一种潜在途径[126].

图7 近干相条件下铁、锰氧化物转化三氯生的可能途径[125]Fig.7 Possible transformation pathways of triclosan by iron or manganese oxides under near dry conditions (after ref.[125])
气态苯酚或氯酚等有机污染物在干燥黏土表面的氧化聚合早在20 世纪初就有相关报道[116-117].2018 年Peng et al[128]和Wang et al[129]报道了在气溶胶中的矿物粉尘表面类似氧化聚合反应的发生,并证明了空气相对湿度对反应的重要影响.他们发现Fe3+‐蒙脱石在低湿条件下能够介导气态2‐氯酚发生二聚反应,蒙脱石中的三价结构铁和伊利石中含有的氧化铁也能介导2‐溴苯酚发生二聚反应形成羟基化的多溴联苯和多溴联苯醚.与Gu et al[121]的研究结果类似,结构铁上的反应在高湿条件下(RH=71%)更易进行.此外,大气环境中多酚类物质还能与矿物粉尘反应生产多聚物[48,130],Ling et al[48]以愈创木酚为模型研究了不同相对湿度下三价铁改性的蒙脱石上的褐碳形成过程,结果表明相对湿度不会影响愈创木酚的聚合途径,但由于水分子对于矿物表面物质扩散和酸度的不同作用,它会影响反应程度和聚合速率.考虑到矿物颗粒是气溶胶中的主要组成成分,且挥发性有机污染物在与矿物颗粒表面相互作用后可以转化为毒性更大的化学物质,在非水饱和相条件下探讨有机污染物在矿物表面的氧化聚合对环境风险的评估具有重要意义.
3.2.2 与多环芳烃反应生成环境持久性自由基(EPFRs)多环芳烃也可以在天然矿物表面通过与过渡态金属离子电子转移产生自由基阳离子,并进一步发生聚合反应[59].Jia et al[131-133]研究表明,多环芳烃通过该途径可以产生环境持久性自由基(EPFRs).实验证明反应相对湿度对PAHs转化及EPFRs 产量有强烈影响.随着RH的增加,自由基中间体产量先升高后降低,当RH>11%时,EPFRs 产量和多环芳烃转化率急剧下降,水的抑制作用归因于多环芳烃和水分子之间对路易斯酸位点的竞争[131].在低湿条件(RH<8%)下,多环芳烃的聚合强度受到多环芳烃种类、黏土矿物类型和黏土矿物中过渡态金属氧化还原电位的强烈影响,蒙脱石上形成的EPFRs 数量比伊利石、高岭石多四个数量级[132-133].
4 总结和展望
本文总结了非水饱和相矿物表面催化有机污染物水解和氧化聚合的最新进展.天然矿物作为重要环境介质可以介导有机污染物发生化学转化,尤其是在干燥土壤表层微生物活性受到抑制时,矿物作用将变得尤其重要.非水饱和相条件下的矿物性质显著区别于水相条件,其在非水饱和相条件下展现的Lewis 和Brönsted 酸性是催化污染物化学转化的重要机制.其中有机污染物的水解被看作是污染物的解毒过程,而氧化聚合一方面是土壤腐殖质的重要天然形成途径,另一方面又反应生成毒性更高的物质(如二噁英),从而对人类健康带来风险.因此在非水饱和相条件对天然矿物化学转化研究至关重要.今后的研究可以从以下方向开展:
(1)目前针对非水饱和相条件下天然矿物介导的污染物催化转化研究仅在蒙脱石、高岭石、铁氧化物等少数典型矿物上进行,还有许多天然矿物如海泡石、二氧化锰、磷灰石、黄铜矿等也亟待研究.
(2)对于非水饱和相中矿物界面对有机物的水解主要选用有机磷农药或氯霉素等易水解有机物作为研究对象,而有关磺胺等其他性质稳定的污染物能否在非水饱和相矿物表面发生同样高效催化水解的研究也具有重要意义.
(3)已有研究证明了黏土矿物在溶液中对雌二醇等内分泌干扰物的低聚反应[134],也应在非水饱和相条件下开展类似研究,为实际条件下土壤或大气颗粒物中反应提供依据.
(4)目前对有机物非水饱和相催化转化仅在单纯矿物上开展,缺乏在实际土壤中的研究.实际土壤含有多种矿物组分且性质更为复杂,且不同地区土壤差异极大.因此应开展非水饱和相条件下实际土壤对污染物催化转化研究,识别主导污染物非生物转化的关键土壤性质和矿物组分,确定土壤非生物和生物降解对污染物转化的贡献度,以揭示环境中污染物自然衰减途径,为修复土壤污染提供新的途径.
