一种超声控制的聚多肽纳米释药系统的制备和表征
2023-06-25张羽殷豪王钦阳
张羽,殷豪,王钦阳
1.温州医科大学附属第二医院育英儿童医院 肿瘤放化疗科,浙江 温州 325027;2.温州医科大学 高等研究院,浙江 温州 325035
当前癌症依然是威胁人类生命安全的一大难题,然而癌症的治疗手段非常有限,化疗仍然是临床上最常用的方式之一。由于化疗药物本身的低选择性与高毒性,导致杀死肿瘤细胞的同时对正常组织细胞也造成严重损伤,更甚者会导致严重的“炎性风暴”,危及患者生命,同时这种低选择性也是产生耐药性的主要原因之一[1]。为了解决这一问题,可以通过借助药物包载将化疗药物递送到肿瘤部位来增加药物的选择性,或是通过外界的刺激在肿瘤部位实现可控的药物释放或药物激活来减少其不良反应[2-4]。目前,常用的刺激方法包括光和超声,其中光刺激具有较低的组织穿透能力,从而限制了它在临床中的使用[5];超声作为一种替代方法,除了具备光刺激的高时空分辨率和非侵入性等优点,它还具有更强的组织穿透力,可以深达15 cm以 上[6]。聚多肽是一种新型的生物医用材料,具有良好的生物活性、生物可降解性和生物相容性,利用聚多肽的自组装机制,可制备出不同尺寸、能适应不同用途的聚多肽药物释放系统[7-8]。本研究构建了一种修饰了声敏剂的聚多肽纳米释药系统,通过聚多肽安全有效地包覆药物,同时通过连接的声敏剂可实现药物的超声可控释放,可有效提高药物的安全性,为化疗药物的精准使用提供一条新的方案。
1 材料和方法
1.1 材料
1.1.1 试剂:分析纯溶剂四氢呋喃购于上海阿拉丁生化科技股份有限公司;分析纯氮-二甲基甲酰胺购于上海阿达玛斯试剂有限公司;苄氧羰基保护赖氨酸和聚乙二醇胺引发剂购自上海乐研公司;三光气和环氧丙烷购自上海麦克林生化科技有限公司;一系列钌络合物以及配体均购自天津希恩斯公司;三氟乙酸和氢溴酸醋酸溶液购自北京百灵威科技有限公司;ATP购于上海安耐吉化学有限公司;萤火虫荧光素酶购于上海颖心实验室;D-荧光素购于上海凯为化学科技有限公司。
1.1.2 仪器:多功能酶标仪(瑞士Tecan公司),纳米粒度仪(奥地利Anton Paar公司),低温冷却循环泵(宁波新芝公司),真空干燥箱(上海一恒科学仪器有限公司),紫外分光光度仪(上海凌析仪器有限公司),超声治疗仪(英国EMS Physio公司),磁力加热搅拌器(德国IKA公司)。
1.2 方法
1.2.1 单体合成:在250 mL圆底烧瓶中,加入磁子,苄氧羰基保护赖氨酸固体2 g,四氢呋喃100 mL,环氧丙烷5 mL,最后快速加入三光气2.1 g,真空硅脂涂抹于瓶口,并快速使用玻璃塞密封并搭配磨口夹避免玻璃塞喷出。反应液初始为固液混合相。反应5 min左右,反应放热,固体逐渐溶解。30 min左右,大部分固体溶解。反应2 h后,反应液为澄清淡绿色溶液。使用棉花过滤得到澄清液体,旋转蒸发旋干除去溶剂,在4 ℃条件下使用50~100 mL分析纯石油醚扩散结晶。结晶完毕后,恢复至室温,使用石油醚/乙酸乙酯=10/1洗涤,抽干,得到为白色固体粉末的单体。
1.2.2 聚多肽合成:将引发剂聚乙二醇胺50 mg加入到50 mL圆底烧瓶中,加入磁子,在氮气保护条件下加入纯化超干氮,氮二甲基甲酰胺2 mL,氮气条件下室温搅拌2 h,引发剂逐渐溶解。在氮气条件下加入之前合成的单体并加入苄氧羰基保护赖氨酸氮羧基内酸酐(N-carboxyanhydrides, NCA),在氮气条件下40 ℃反应4 h,固体全部溶解。使用薄层层析色谱监控显示单体反应完全并且溶液澄清。直接将全部反应液透析24 h,析出固体粉末,离心冻干,得到白色粉末的聚合物。取50 mL圆底烧瓶,加入磁子,聚合物10 mg,0 ℃条件下逐滴加入三氟乙酸2 mL,搅拌10 min聚合物完全溶解,然后逐滴加入0.5 mL氢溴酸乙酸溶液,0 ℃反应2 h,此时溶液为淡黄色澄清溶液,并产生少量固体不溶物。 0 ℃下快速加入乙醚50 mL,白色固体析出,乙醚洗涤2次,真空干燥,得到淡黄色胶状物的聚多肽。
1.2.3 声敏剂合成:取250 mL圆底烧瓶,加入磁 子,200 mg二氯二联吡啶Ru络合物,150 mg联吡啶二羧酸,200 mg碳酸氢钠,32 mL甲醇和8 mL去离子水。架球形冷凝管,80 ℃条件回流16 h。反应液从初始紫色变成深黄色。在0 ℃条件下,逐滴滴入浓硫酸,使得反应体系的pH为4.4,并且在0 ℃下搅拌2 h,此时析出固体。使用砂芯漏斗,过滤固体,使用8 mL甲醇洗涤固体,合并滤液。在0 ℃下,加入12.5 mL六氟磷酸钠水溶液(2.5 g六氟磷酸钠溶于12.5 mL去离子水中),0 ℃搅拌2 h,此时产生深棕色沉淀。离心,弃去上清液,冻干得到羧酸配体固体产物。取230 mg二环己基碳二亚胺,120 mg 氮羟基琥珀酰亚胺,使用2 mL N, N-二甲基甲酰胺溶解,0 ℃条件下冷却,加入190 mg冻干产物,0 ℃ 条件下搅拌30 min然后室温搅拌5 h。离心取上清液,得到声敏剂材料的N, N-二甲基甲酰胺溶液。使用紫外可见光分光光度计测量,得到吸光度,并根据文献报道的吸光系数计算浓度[9]。
1.2.4 超声响应型聚多肽纳米材料合成:取50 mL圆底烧瓶,加入磁子,聚多肽5 mg,三乙胺2 mL,合成的声敏剂材料2 mL,氮气避光条件下,加入纯化N, N二甲基甲酰胺5 mL。室温反应16 h,溶液呈现深棕色。避光条件下,反应液直接透析24 h,冻干,得到深黄色固体状的超声响应型聚多肽(见图1)。
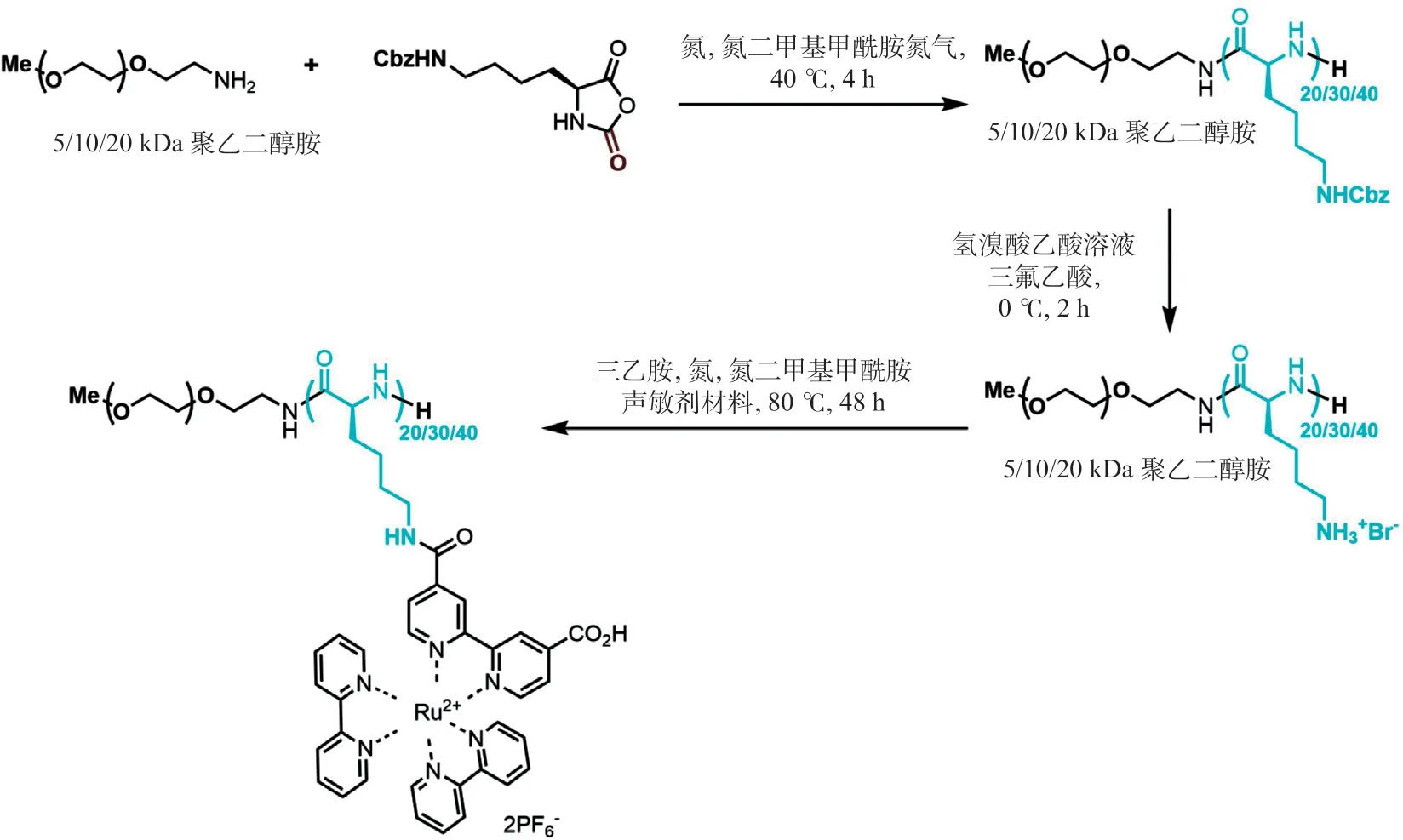
图1 超声控制的聚多肽纳米系统的合成路径
1.2.5 超声响应型聚多肽纳米释药系统合成:取20 μL含超声响应型聚多肽的N, N二甲基甲酰胺溶液(10 mg/mL),与20 μL ATP的磷酸缓冲溶液水溶液(20 mg/mL)混合3 min,直接加入2 mL酸缓冲溶液混合,超滤去除溶剂,反复使用酸缓冲溶液3 mL 超滤3次,最后获得包载ATP的浓度为0.1 mg/mL的声敏剂聚多肽纳米颗粒的酸缓冲溶液(见图2)。

图2 超声控制的聚多肽纳米释药系统的示意图
1.2.6 超声响应型聚多肽纳米释药系统相关性能表征:①各组分核磁共振检测及材料形貌表征。使用Bruker AvanceNEO 600 MHz核磁共振波谱仪检测超声响应型聚多肽纳米释药系统的单体和声敏剂修饰的聚合物,使用透射电子显微镜(FEI Tecnai 12)检测超声响应型聚多肽纳米释药系统。②粒径及zeta电位检测。使用纳米粒度仪检测超声响应型聚多肽纳米释药系统超声前后的粒径,以及包载ATP前后和超声前后的zeta电位。③ATP的包载率及超声释放效率[10-11]。于96孔板中分别加入 0.05 mol/L Tris缓冲溶液(pH 7.8)、萤火虫荧光素酶、底物和ATP,利用酶标仪检测溶液光强度,得到ATP释放的标准曲线(见图3)。在合成包载ATP纳米颗粒前后,分别对ATP母液以及纳米颗粒包载后的混合溶液进行萤火虫荧光素酶ATP检测,包载ATP效率的计算公式为:包载率=(ATP母液中ATP量-包载后的ATP量)/(ATP母液中ATP量)。超声前使用萤火虫荧光素酶检测样品母液的ATP浓度,在1 Wcm-2, 1:4脉冲条件下超声10 min后再次检测溶液的ATP含量,ATP释放率计算公式为:释放率=(超声后的ATP量-超声前ATP量)/(ATP包载总量)。④稳定性检测。在未超声的情况下即pH为7.4,温度为37 ℃的PBS溶液中,通过检测24 h内超声响应型聚多肽纳米释药系统ATP含量和粒径的变化,以表征其稳定性。⑤细胞毒性检测。将处于生长对数期的293T细胞铺入96孔板,每孔200 μL,10000个细胞/孔,放入细胞培养中6 h使其贴壁;加入不同浓度的超声响应型聚多肽纳米释药系统,继续培养24 h; 小心吸去上清液,加入100 μL含10% MTT溶液的新鲜培养基,继续培养4 h;小心吸去上清液,每孔加入110 μL的DMSO,并于摇床上低速振荡10 min;使用酶标仪检测490 nm处的吸光值。每组设定3个复孔。⑥超声释放条件优化。在不同的超声功率下对超声响应型聚多肽纳米释药系统1:4脉冲超声 10 min,检测其ATP释放率;并使用上述MTT法检测不同超声条件对293T的细胞毒性。对超声响应型聚多肽纳米释药系统在1 Wcm-2,1:4脉冲条件下超声不同的时间,检测其ATP释放率。

图3 利用萤火虫荧光素酶检测ATP释放的标准曲线
1.3 统计学处理方法采用GraphPad Prism 8.0软件对数据进行统计学分析。计量资料以±s表 示,两组间比较采用t检验;多组间比较采用单因素方差分析,组间两两比较用LSD-t检验。P<0.05为差异有统计学意义。
2 结果
2.1 各组分核磁共振检测及材料形貌表征通过对合成的单体及声敏剂修饰的聚多肽进行核磁共振检测表明已成功合成相关化合物,同时通过透析等手段可有效保障合成的化合物的纯度,有效降低相关残留化合物对细胞的毒性(见图4和图5)。同时对包载ATP的超声控制的聚多肽纳米释药系统进行透射电镜分析,其形态为实心圆球形,且粒径较为均一(见图6)。

图4 单体核磁共振检测结果
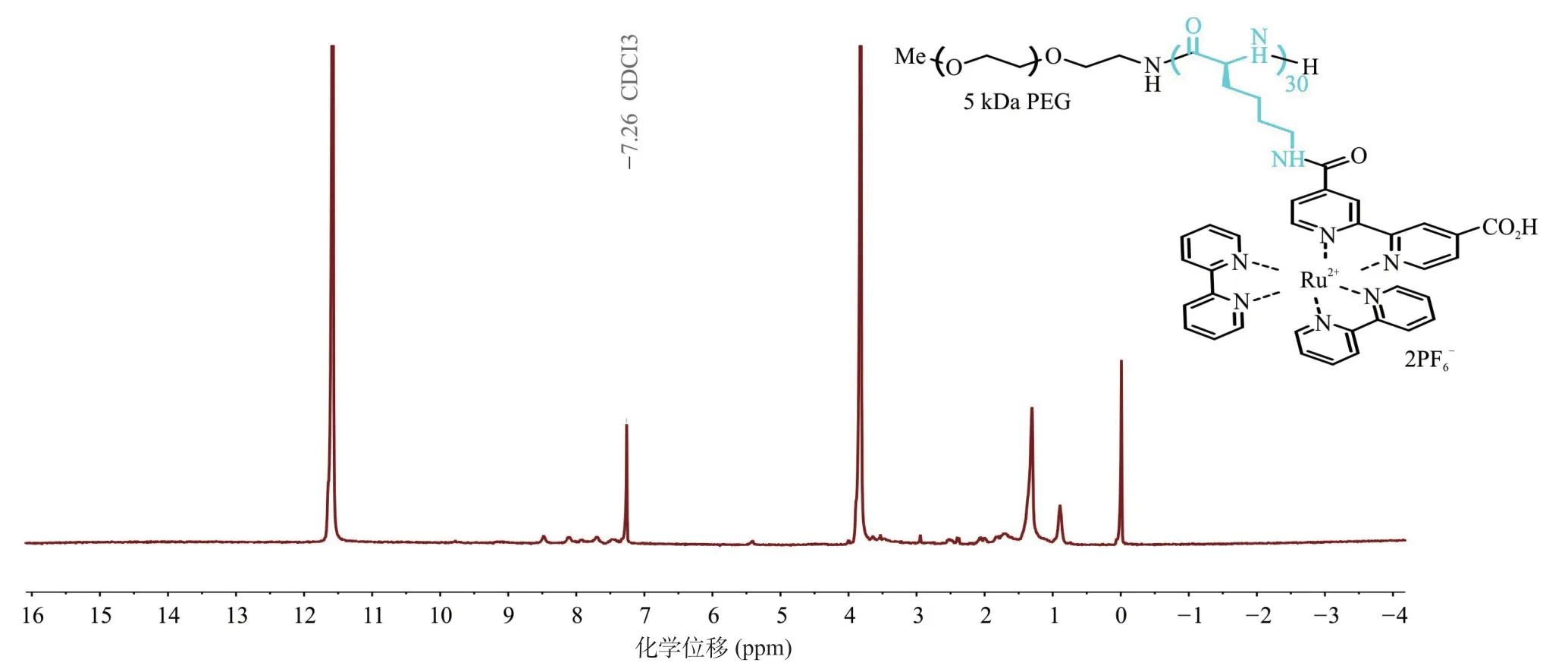
图5 声敏剂修饰的聚多肽核磁共振检测结果

图6 10K(1:30)超声响应型聚多肽纳米释药系统的透射电镜图
2.2 包载率通过分别加入5K、10K和20K的引发剂和聚合度20、聚合度30和聚合度40的聚多肽以合成8种不同的超声控制的聚多肽纳米释药系统(见表1)。对于5K引发剂来说,聚合度20的超声响应型聚多肽纳米释药系统的包载率显著高于聚合度30的包载率(P<0.05)。但当聚合度上升到40的时候,由于聚合物大量沉降,而且颗粒巨大无法溶解,因此无法表征。对于引发剂10K与20K的超声响应型聚多肽纳米释药系统,其包载率显著高于5K引发剂聚合度30的超声响应型聚多肽纳米释药系统(P<0.05),但是显著低于5K引发剂聚合度20的超声响应型聚多肽纳米释药系统(P<0.05)。引发剂10K 与20K的超声响应型聚多肽纳米释药系统形成的包载后粒径明显大于5K引发剂的超声响应型聚多肽纳米释药系统(P<0.05);10K引发剂聚合度30的超声响应型聚多肽纳米释药系统粒径最大,可达129 nm, 显著高于5K引发剂、20K引发剂以及10K引发剂聚合度20、10K引发剂聚合度40的超声响应型聚多肽纳米释药系统粒径(P<0.05)。利用紫外可见光在最大激发波长扫描和最大发射波长扫描表征声敏剂材料的连接,单纯的聚多肽没有紫外可见光的吸收和发射,而当声敏剂修饰后出现了与单纯声敏剂类似的吸收和发射,表明通过本研究合成方法可成功将声敏剂修饰到聚多肽上(见图7)。

表1 不同引发剂和聚合度的超声响应型聚多肽纳米释药系统表征

图7 10K(1:30)超声响应型聚多肽纳米释药系统各组分的紫外-可见吸收光谱
2.3 稳定性在未超声、pH为7.4、温度为37 ℃的PBS溶液中,测定超声控制的聚多肽纳米释药系统ATP含量变化以及粒径变化表征其稳定性。24 h内ATP含量和粒径与0 h比差异均无统计学意义(P>0.05);同时超滤前后的粒径差异也无统计学意义(P>0.05)。提示超声控制的聚多肽纳米释药系统具有良好的载药稳定性(见图8和图9)。

图8 不同时间下超声控制的聚多肽纳米释药系统ATP含量和粒径变化
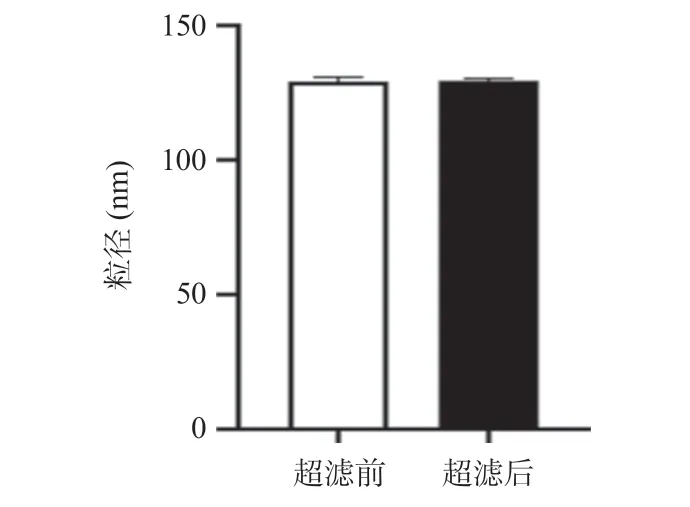
图9 超滤对于超声响应型聚多肽纳米释药系统粒径的影响
2.4 超声响应性释放率7种聚多肽纳米释药系统在超声后均显示出了不同程度的粒径变小,超声过程中纳米颗粒发生解聚,在超声停止后,解聚的纳米材料再次发生聚集,导致粒径变小。10K引发剂聚合度20和10K引发剂聚合度40的聚多肽纳米释药系统在超声前后粒径变化仅为2~3 nm,显著低于其他5种聚多肽纳米释药系统在超声前后可达10 nm及以上的粒径变化,差异有统计学意义(P<0.05)。在超声过程当中,纳米颗粒解聚可以导致包载的ATP发生释放,7种聚多肽纳米释药系统在超声后显示出了不同程度的ATP释放,10K引发剂聚合度30的聚多肽纳米释药系统在超声后释放率最高(可达15%),显著高于其他6种聚多肽纳米释药系统的释放率,差异有统计学意义(P<0.05)。见表1。10K引发剂聚合度30 的聚多肽纳米释药系统超声后与超声前比粒径减小,差异有统计学意义(P<0.05),溶液中ATP含量增多,差异有统计学意义(P<0.05)。ATP包载前后和超声前后材料zeta电位变化显示,包载ATP后材料的zeta电位变小,差异有统计学意义(P<0.05),而超声前后zeta电位差异无统计学意义(P>0.05)。提示材料成功包载了ATP,聚多肽在超声后重新自组装保障了药物的缓释及可控性(见图10和图11)。

图10 超声前后10K(1:30)聚多肽纳米释药系统的粒径变化和溶液中ATP含量的变化
图11 10K(1:30)聚多肽纳米释药系统在ATP包载前后和超声前后的zeta电位变化
2.5 细胞毒性不同浓度组的超声响应型聚多肽纳米释药系统细胞活性与0 μg/mL组比差异均无统计学意义(P>0.05),超声响应型聚多肽纳米释药系统浓度达到10 μg/mL时对293T细胞的细胞毒性依然较低(见图12)。提示超声响应型聚多肽纳米释药系统其本身毒性较低,具有较好的生物安全性。

图12 超声响应型聚多肽纳米释药系统的细胞毒性
2.6 超声释放条件优化在相同超声时间下,随着超声功率的增加,聚多肽纳米释药系统ATP释放率也逐渐增加,1.0 Wcm-2组与0.5 Wcm-2组、1.5 Wcm-2组与1.0 Wcm-2组、2.5 Wcm-2组与2.0 Wcm-2组比差异均有统计学意义(均P<0.05)。随着超声功率的增加,细胞活性也逐渐下降,1.5 Wcm-2组与 1.0 Wcm-2组、2.0 Wcm-2组与1.5 Wcm-2组、2.5 Wcm-2组与2.0 Wcm-2组比差异均有统计学意义(均P<0.05)。在相同超声功率下,随着超声时间的增加,聚多肽纳米释药系统ATP释放率也逐渐增加,10 min 组与1~8 min组比差异均有统计学意义(均P<0.05)。超声功率为1.0 Wcm-2、超声时间为10 min时超声控制的聚多肽纳米释药系统具有较好的药物释放效率,且对细胞损伤也最小,具有更好的安全性(见图13和图14)。

图13 超声功率对聚多肽纳米释药系统ATP释放率及对293T细胞的损伤的影响

图14 1 Wcm-2超声功率下超声时间对材料ATP释放效率的影响
3 讨论
纳米药物递送系统作为一种新型的药物释放方法受到广泛关注和研究,相比于传统的释药方式具有可以提高药物的稳定性、减少药物的降解、减轻药物的全身不良反应,并且可以提高靶点的药物浓度和药物的生物利用度等优势。目前,研究较多的纳米药物递送系统主要包括脂质体纳米递送系统、聚合物纳米递送系统和无机材料递送系统[12]。其中,脂质体纳米递送系统的制备成本相对较高,并且存在长期贮存不稳定、易泄露,其在体内循环稳定性也较低、易导致药物的提前释放。且其制备工艺不成熟,存在不同批次间差异较大、粒径难以控制、及有机溶剂残留等问题,这些不足都极大限制了脂质体释药系统的进一步应用。无机纳米递送系统由于其溶解性低和潜在的毒性,只有少数载体获得批准[13]。聚多肽纳米释药系统由于其良好的生物相容性、生物降解性、水溶性和贮存稳定性受到了更多的关注。以往研究发现,多种因素可以影响聚多肽纳米递药系统的响应性,包括化学键的键 能[14]、聚合物的分子量和聚合度[15-16]、分子量的分布[17]、聚合物的形状和结构以及聚合物的组装等[18-20]。所以如何通过优化不同组分提高聚多肽纳米释药系统的性能,同时对其可控释药方式进行探究具有重要意义。
本研究中设计的聚多肽纳米释药系统,以不同引发剂和聚合度的聚多肽纳米材料为载体,并引入声敏剂使其具备超声响应性,有效地提高了聚多肽纳米释药系统的稳定性以及可控释药性能。本研究对合成聚多肽的引发剂的分子量及其聚合度的影响进行了探索,发现使用5K引发剂时,聚合度20的超声响应型聚多肽纳米释药系统的包载率明显高于聚合度30 的超声响应型聚多肽纳米释药系统的包载率。但当聚合度上升到40的时候,聚合物颗粒巨大,无法溶解且大量沉降,这是因为聚赖氨酸部分在声敏剂材料修饰后疏水性急剧上升,同时自身比重过高,导致整个聚合物的溶解性下降。因此,聚合度30的包载率低于聚合度20很有可能是因为聚合度30的声敏剂聚合物材料溶解性较差导致。对于引发剂10K与20K的声敏剂聚合物材料,在相同聚合度的条件下,10K的聚合物粒径要略微大于20K,提示亲水性更强的聚合物更利于形成小粒径的纳米颗粒。但其粒径在超声前后的变化规律不太明显,这也符合聚合物中的亲疏水比例对聚合物大小以及粒径影响的经验情况:如聚合物的组装大小、形态以及比例,但其影响因素到目前没有一个很明确的理论。10K引发剂聚合度30的聚多肽纳米释药系统不仅具有较好的包载率,超声响应的药物释放率也最高,为最合适的超声控制的聚多肽纳米释药系统。
本研究制备的超声响应型的聚多肽纳米释药系统,具有良好的载药效率、稳定性和超声可控性。通过借助于超声引起的机械效应,在超声部位实现可控的药物释放;且由于超声具备优异的组织穿透力,可以穿透深层组织和大多数的实体瘤,将有望实现体内精准的药物释放。因此,本研究制备的超声响应型聚多肽纳米释药系统为后续超声控制的体内释药系统研究提供了一个新方向。
