乙酸基+O2及其分解异构化反应速率常数计算和不确定度分析
2023-06-15殷阁媛肖波胡二江黄佐华
殷阁媛,肖波,胡二江,黄佐华
(西安交通大学动力工程多相流国家重点实验室,710049,西安)
大气中的羧酸加强了大气、硫酸和硝酸的酸性,是导致酸性沉降(酸雨)的主要成因之一[1]。以往的研究证明酸性污染物主要来源是交通运输设备的排放,通过含氧燃料的氧化和热解过程形成[2]。明确羧酸在发动机内的形成路径和后续消耗反应机制,从燃烧源头控制是抑制羧酸生成并促进其消耗,是降低排放最有效的手段之一。生物质柴油热解释放的焦油中含氧物种非常丰富,其中乙酸是其挥发分的主要酸性成分,通常可以作为生物柴油替代物模型的典型组分[3-4],因此研究乙酸的消耗路径,构建准确的化学反应动力学模型是非常必要的[5]。
Gasnot等[6]类比了碳氢燃料甲基脱氢反应,获得了乙酸甲基脱氢的速率常数,发展了乙酸的动力学模型。Leplat等[7]在模型中添加了羧酸基脱氢反应簇的速率常数,该反应簇类比了醇类的脱氢反应。自由基与O2的反应在中间温度范围以及高压条件下对着火非常重要,该反应能够通过消耗O2,产生活化自由基RO2。RO2后续会碰撞稳定,异构化或者直接分解,从而产生活泼的OH基,促进整体反应活性。Burke等[8]开展的敏感性分析中甲基乙烯基基与O2反应的敏感性系数高达0.2,对丙烯燃烧活性非常重要。Goldsmith等[9-10]针对烷烃自由基与O2反应系统开展了高精度的量子化学计算,强调了该类反应的重要性。然而,酸类自由基相关反应研究非常稀少。自由基异构化分解反应对高温条件的着火动力学意义重大,包括烷烃、烯烃和醇类的路径分析均[11-13]证明在高温条件下脱氢后自由基主要的消耗路径是其异构化和分解反应。在我们之前关于甲酸的研究中表面自由基与O2的反应及其自身异构化和分解几乎消耗了100%的甲酸基。Christensen和 Konnov[14]发表的化学反应动力学模型中缺少乙酸基与O2反应,而其自身分解逆反应速率常数被估计为1014cm3·mol-1·s-1。
因此,为了获得乙酸基(CH2CO(O)H)上述反应簇准确的温度压力依赖的速率常数,明晰该反应类型在高低温反应动力学中的竞争关系及其对乙酸消耗的作用,并为酸类模型的构建提供指导,开展了高精度的量子化学计算及全面的不确定度分析。所有重要的反应如下
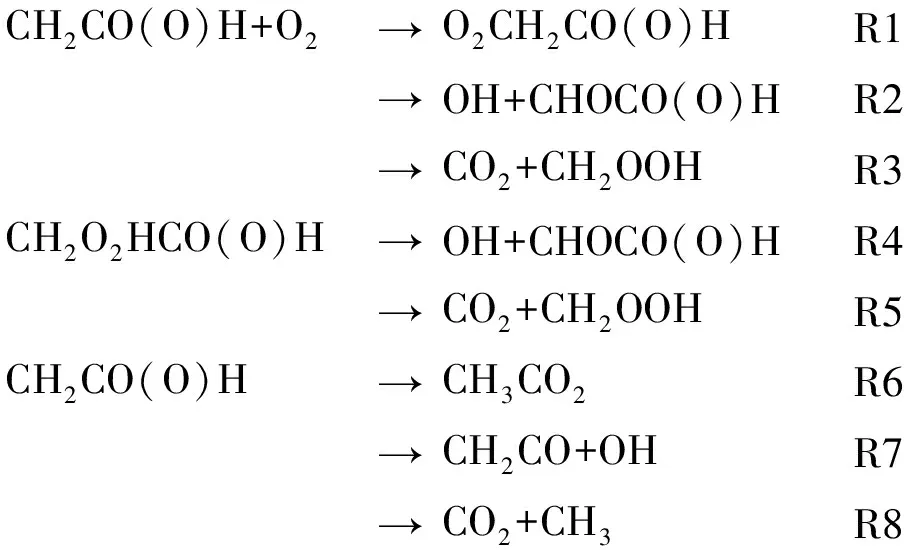
CH2CO(O)H+O2→O2CH2CO(O)HR1→OH+CHOCO(O)HR2→CO2+CH2OOHR3CH2O2HCO(O)H →OH+CHOCO(O)HR4→CO2+CH2OOHR5 CH2CO(O)H→CH3CO2R6→CH2CO+OHR7→CO2+CH3R8
1 计算方法
1.1 电子结构计算方法
计算速率常数需要准确的分子结构,但该项计算需要耗费大量的计算资源和时间,因此,选择合适的方法至关重要。本文采用密度泛函理论(DFT)中的B2PLYPD3方法和Dunning相关一致性基组(cc-pVnZ系列)cc-pVTZ[15-16]对反应物、产物以及过渡态(TS)的分子结构进行了优化,并对它们的频率进行了计算。基于DFT优化后的结构,采用耦合簇方法CCSD(T)-F12和cc-pVTZ-F12[17]基组计算单点能。所有的过渡态均采用路径分析扫描,确保过渡态连接了正确的反应物和产物。对于无势垒的反应,采用二阶多参考微扰方法(CASPT2)和cc-pVTZ基组优化结构,并将基组外推至全基组计算单点能,采用的公式[18]为
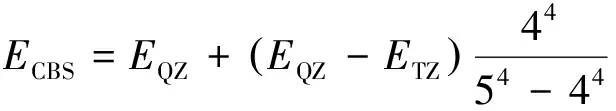
(1)
式中:ECBS为全基组能量外推结果;EQZ为cc-pVQZ基组能量;ETZ为cc-pVTZ基组能量。
采用DFT的计算在Gaussian 09[19]中进行,采用CCSD(T)和CASPT2的计算在Molpro[20]中进行。
1.2 可变反应坐标系过渡态理论[21-22]
CH2CO(O)H与O2自由基反应生成O2CH2CO(O)H为无势垒反应。因此,在VaReCoF软件[23-24]中,采用可变反应坐标系过渡态理论计算这些反应通道的速率常数。利用该理论计算过渡态的配分函数时,将过渡态的自由度分解为守恒自由度和过渡自由度,守恒自由度是指两个片段的内部自由度而过渡自由度则是二者之间相对耦合的非简谐自由度。对于后者,利用蒙特卡罗方法对大量任意取向的构型进行经典相空间积分,同时利用两种方法,计算了反应的最小能量路径。第一条仅固定反应坐标,体系中守恒的自由度进行放开优化;第二条固定反应坐标,同时固定守恒的自由度,亦即保持两反应物(通常为自由基)各自的构型,只优化两者之间的相对运动模式,计算过程中二者之间的差值作为修正值,同时对于基组采用同样的方法进行修正。
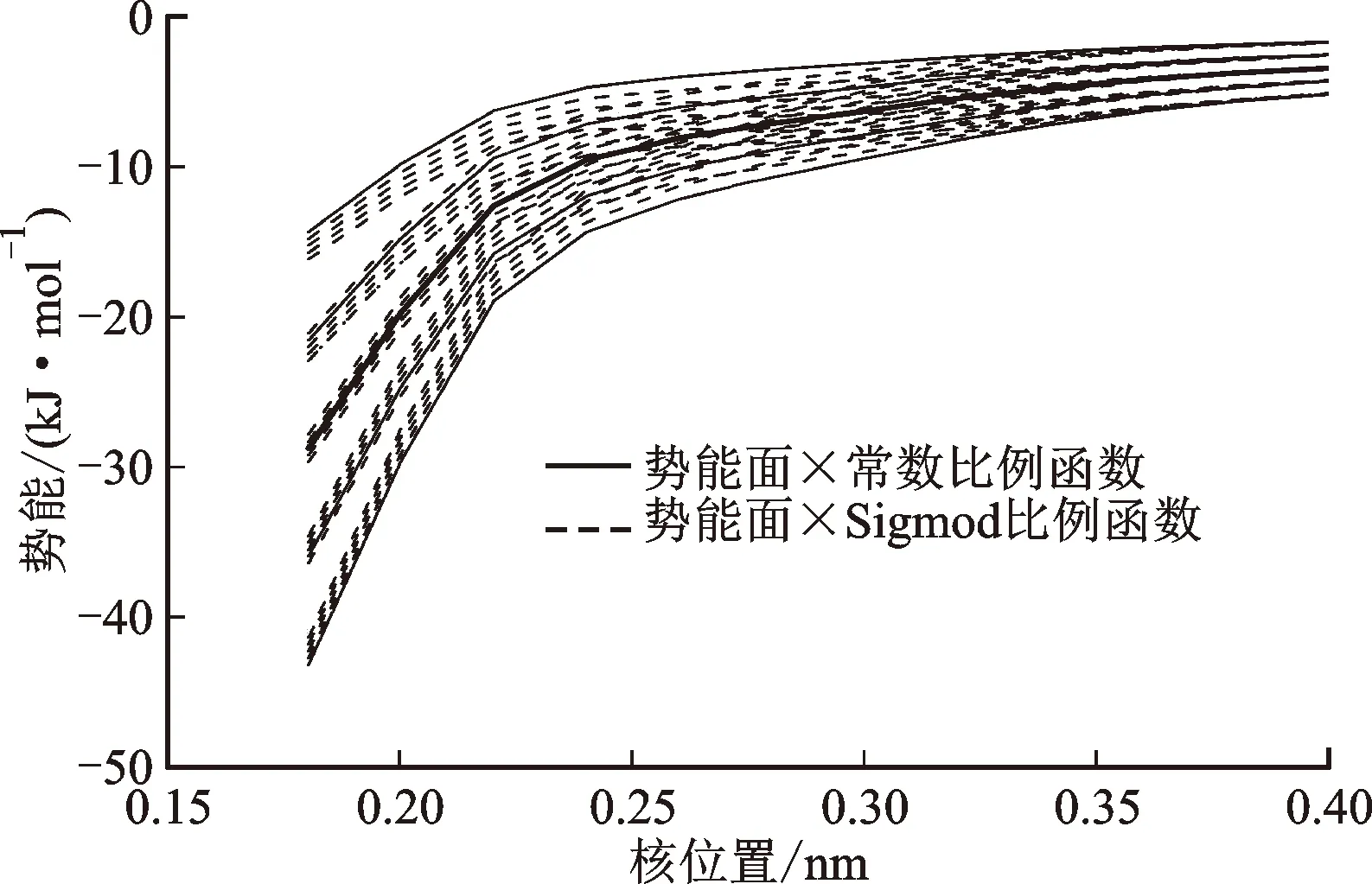
本文开展了有势垒和无势垒反应全面的不确定度分析,针对无势垒反应,利用比例函数f(r)构建反应势能面50%和25%的不确定度区间,如图1所示,采用了常数比例函数和Sigmoid比例函数。针对Sigmoid比例函数,设置不确定度变化最快出现的位置为势能面瓶颈位置,负值和正值分别代表势能面相对于平均势能面缩小或者增加,势能面V(r)与比例函数f(r)的关系为V(r)=Vaverage(1+f(r)),Vaverage为势能面均值。将不确定区间内的势能面簇转化为一维的修正值,加入可变坐标系速率常数计算过程,以获得无势垒反应的速率常数不确定度。
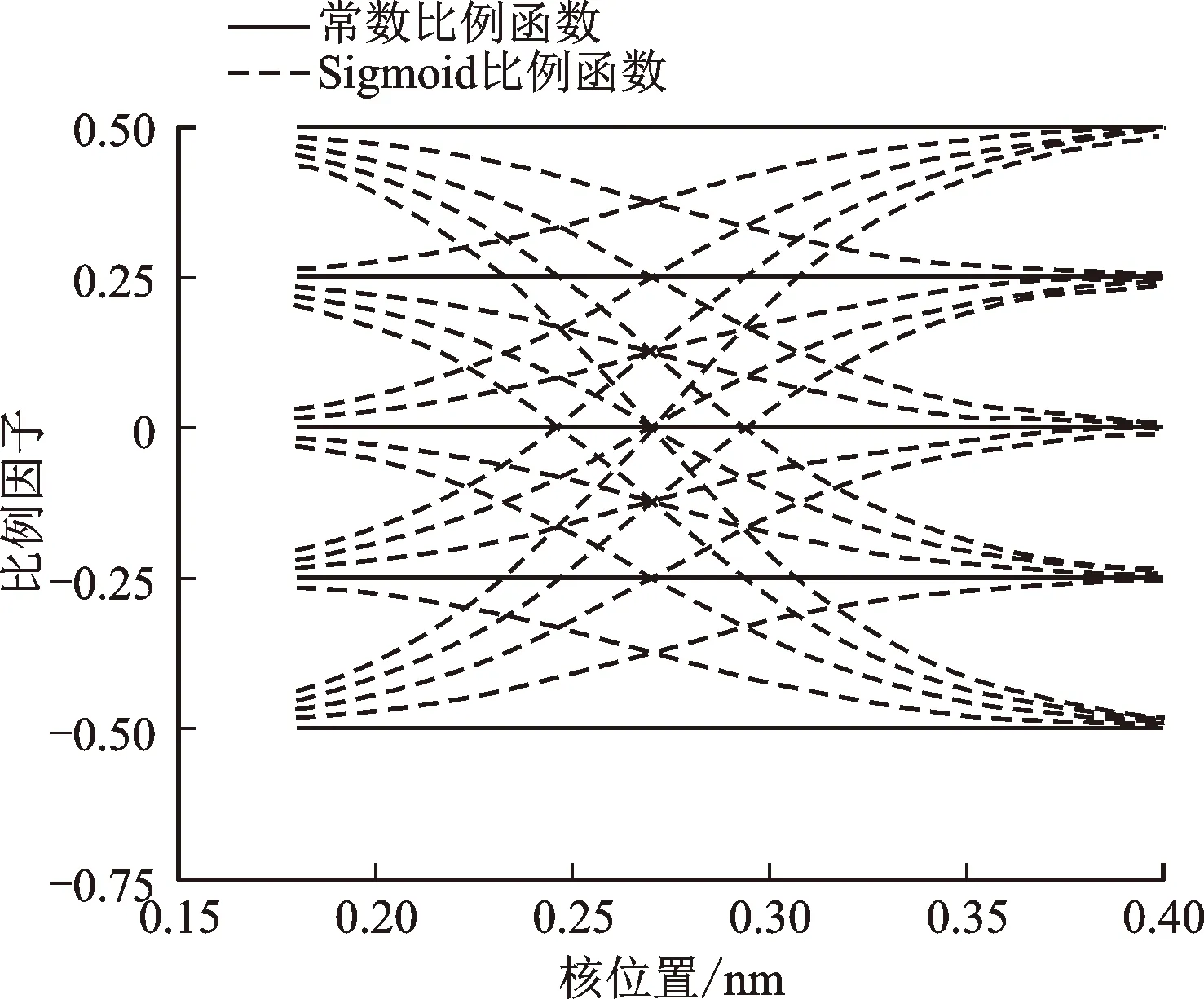
图1 势能面上的一系列比例函数
1.3 RRKM和主方程求解
温度和压力相关的速率常数通过求解RRKM和主方程的软件MESS[20]计算得到。所有物种(包括反应物、产物和过渡态)的低频内转动均视为阻尼内转动。通过DFT结合3-zeta基组(M06-2X/6-311g(d, p))扫描转动势能面,结果拟合为傅里叶函数带入配分函数的计算。隧道效应通过非对称的Eckart假设计算。主方程中的能量传递采用单指数下降模型,其中的平均碰撞能量转移参数〈ΔEdown〉表达为温度的函数A0.8(T/298),对于氮气,A选为400 cm-1。反应物与载气分子之间的相互作用力以兰纳-琼斯(Lennard-Jones, L-J)势函数表征,CH2CO2H[9]+O2参数:σ=0.442 nm,ε=292 cm-1;N2参数:σ=0.420 nm,ε=351 cm-1。该类反应的误差来源主要为能垒、频率以及碰撞参数,本文利用蒙特卡罗方法研究能垒、频率以及碰撞参数耦合作用下不确定度的传递机制,其中能垒、虚频、低频以及碰撞参数的不确定度分别为4.18 kJ·mol-1、20%、20%以及50%。
2 结果与讨论
2.1 反应势能面

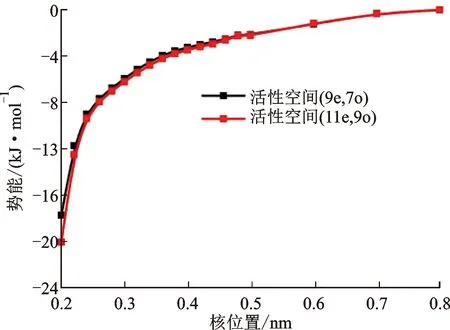
图2 采用不同活性空间获得无势垒反应CH2CO(O)H+O2反应势能面
(a)基于图1获得势能面簇
乙酸基与O2反应及其自分解和异构化反应的单点能列于表1、表2,反应势能面如图4所示。在图4(a)中,O2首先加成到乙烯基的C原子上构成RO2的顺反异构体分别记为RO2(顺)和RO2(反),其中RO2(顺)是所有构象里的能量最低的,且与RO2(反)极易发生异构化反应,在燃烧对应的温度区间里,二者的差别可以忽略。RO2(顺)后续会通过过渡态TS2分解为CHOCO(O)H+OH(P2),势垒高为178.1 kJ·mol-1。RO2(反)则会通过TS3分解生成CH2O2H+CO2(P1),势垒相对较低为77.3 kJ·mol-1,低于反应物,因此由于碰撞而化学活化的反应物能够越过势阱W2,生成P1。
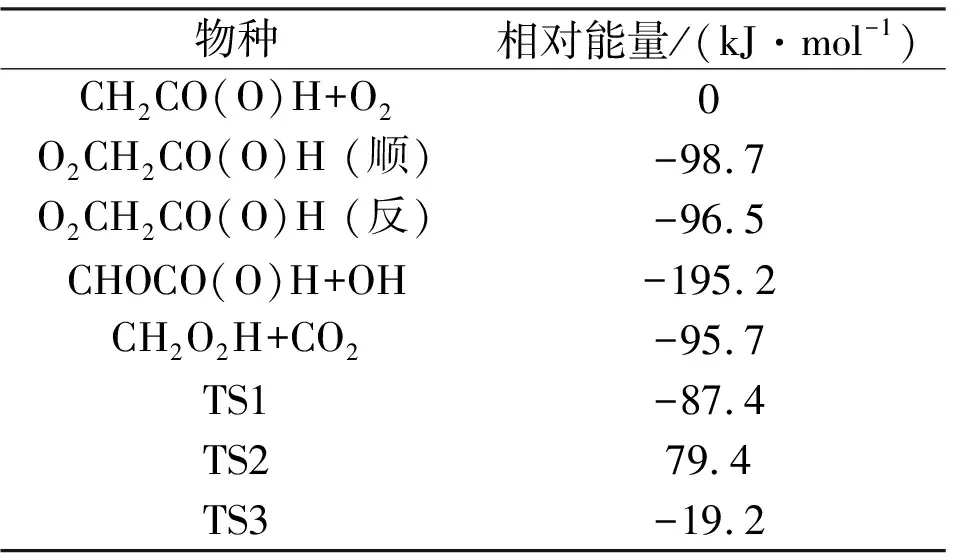
表1 CH2CO(O)H与O2体系的单点能
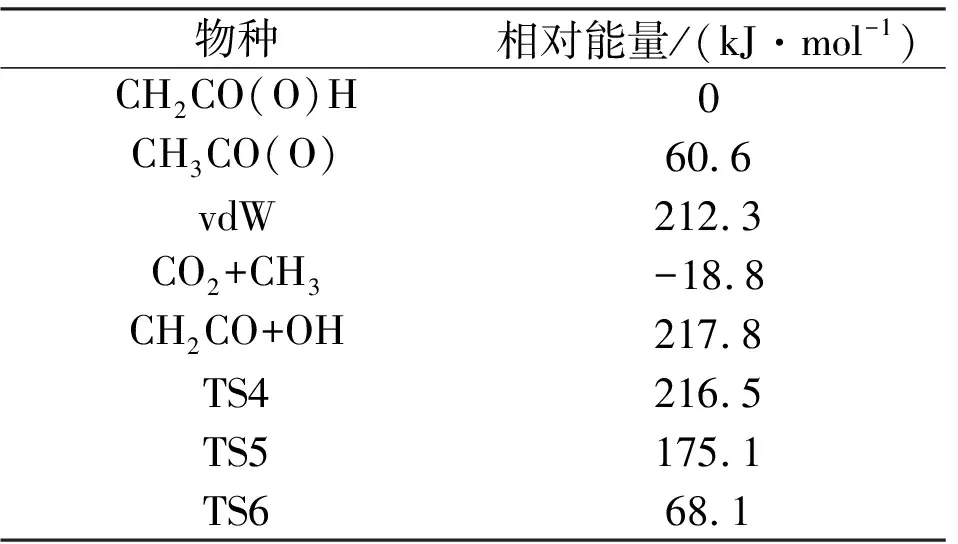
表2 CH2CO(O)H异构化及分解体系的单点能
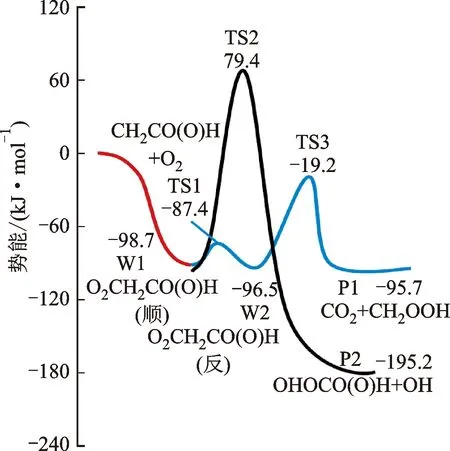
(a)CH2CO(O)H与O2反应
CH2CO2H自分解和异构化反应势能面如图4(b)所示,CH2CO2H的OH键直接断键可以生成CH2CO+OH(P3),由于范德华作用力在反应路径生成了络合物vdW。CH2CO2H同样可以异构化生成CH3CO2,而CH3CO2会通过很低的势垒,7.5 kJ·mol-1,分解生成CO2+CH3(P4)。
2.2 反应速率常数
2.2.1 CH2CO(O)H与O2反应
在不同压力下,CH2CO(O)H与O2加成反应及后续分解反应的速率常数如图5所示。CHOCO(O)H+OH产物通道的速率常数相比于其他产物低2个数量级以上,在整个反应体系中并不重要,因此并未表示在图5中。当压力为0.001 MPa、温度超过700 K时,超过99%的化学活化的O2CH2CO(O)H自由基直接分解为CH2O2H+CO2。随着压力升高,温度降低,化学活化的O2CH2CO(O)H碰撞稳定后生成O2CH2CO(O)H,O2CH2CO(O)H则在后续反应中分解为CH2O2H+CO2。当压力为10 MPa时,在整个温度区间主要的生成O2CH2CO(O)H。
2.2.2 CH2CO2H分解与异构化反应
自由基异构化分解反应对高温条件的着火动力学意义重大,包括烷烃、烯烃和醇类的路径分析均证明,在高温条件下脱氢后,自由基主要的消耗路径是其异构化和分解反应,因此开展了乙酸自由基异构化分解反应速率常数计算,得到的结果如图6所示。当温度低于800 K时,CO2+CH3反应通道占据主导地位,因为该反应的势垒比其他通道低100 kJ·mol-1,反应速率常数大4个数量级以上。随着温度升高,CH2CO+OH的通道开始变得更加重要,可以与CO2+CH3反应通道竞争。
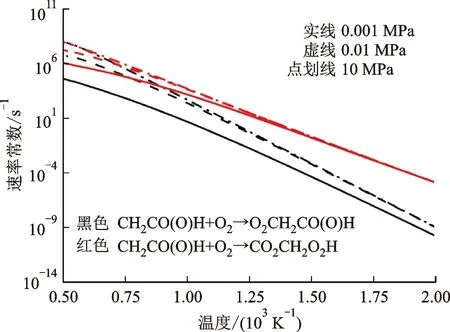
图6 CH2CO(O)H分解和异构化反应速率常数
本文计算的OH+CH2CO→CH2CO(O)H反应速率常数与Christensen模型结果进行对比,同时选择文献[25]相似结构的反应OH+CO→HOCO速率常数进行验证,结果见图7。原模型中该反应的速率常数仅被粗略的估计为4×1012cm3·mol-1·s-1。本文计算的速率常数与文献[25]甲酸基相关反应速率常数在常压下吻合良好,证明了本工作的准确性。本文计算得到的所有压力温度依赖的速率常数见表3。
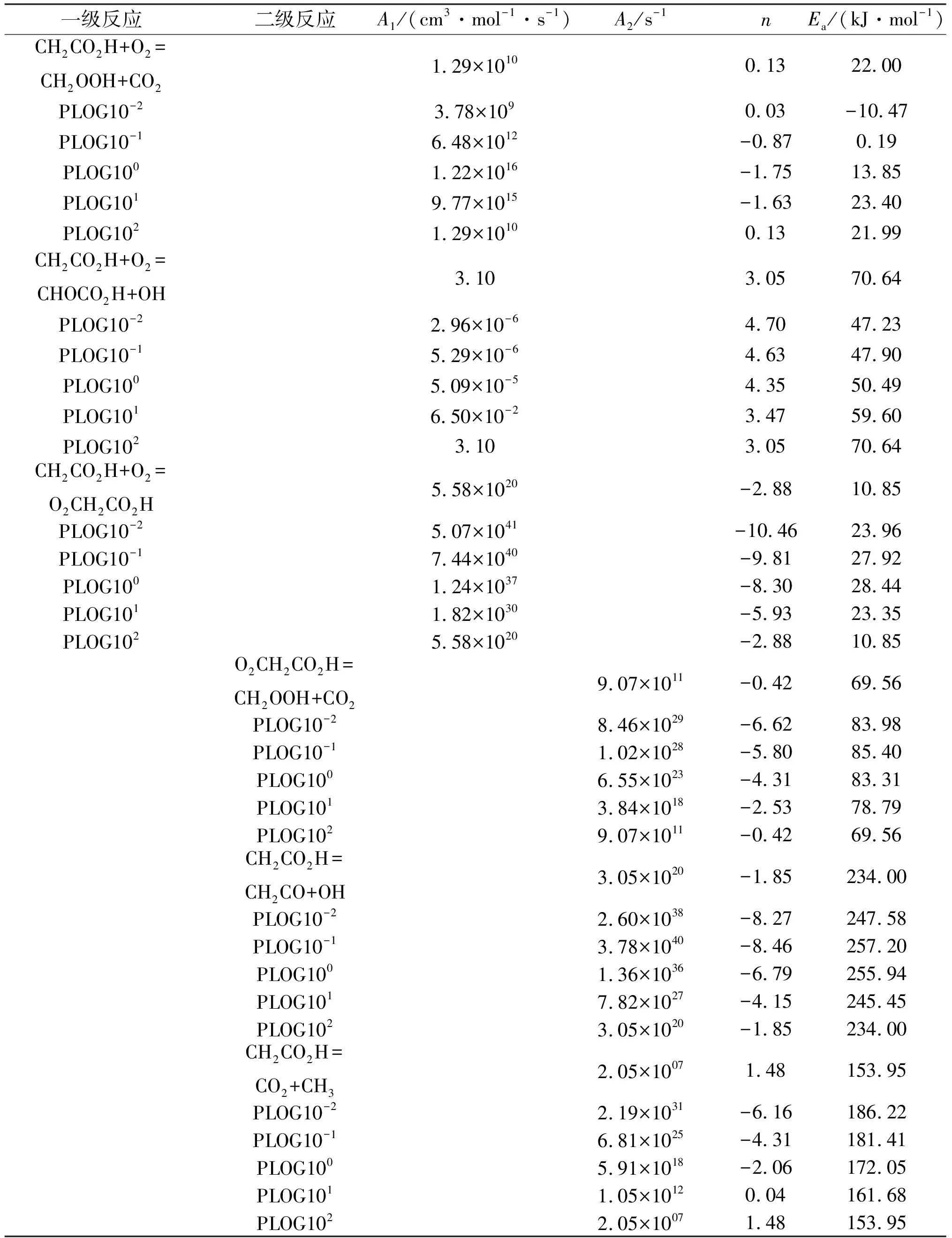
表3 计算得到的反应速率常数
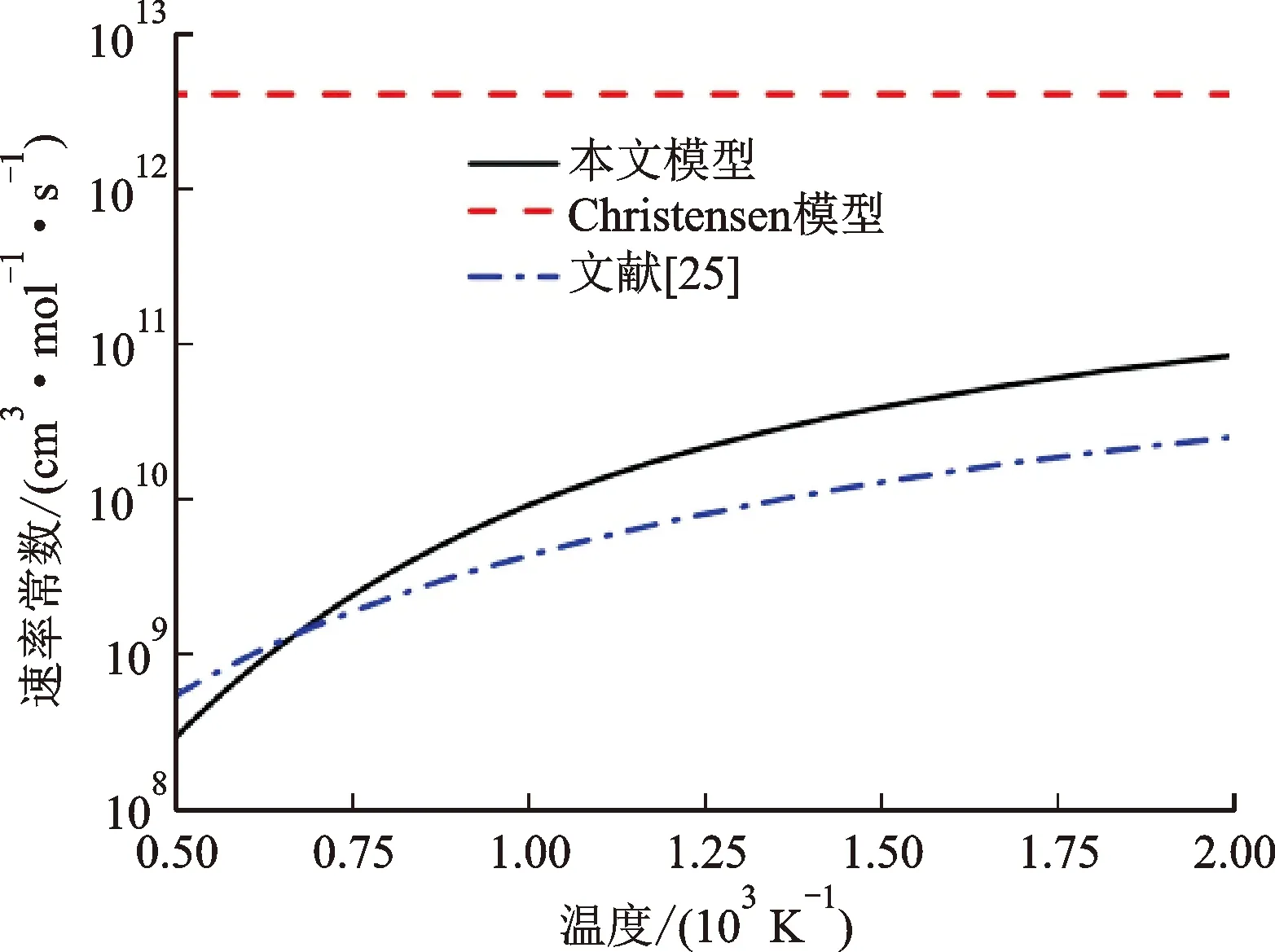
图7 本文计算结果与Christensen模型、文献[25]结果对比
2.3 反应速率常数不确定度分析
通过图1构建的比例函数,计算得到了无势垒反应的势能面50%和25%不确定度区间。将其转化为1维的修正参数带入到速率常数进行计算,结果如图8所示。整个势能面增加25%和50%会使速率常数下降至50%和25%,相反的势能面降低25%和50%会使速率常数增加2或者4倍。
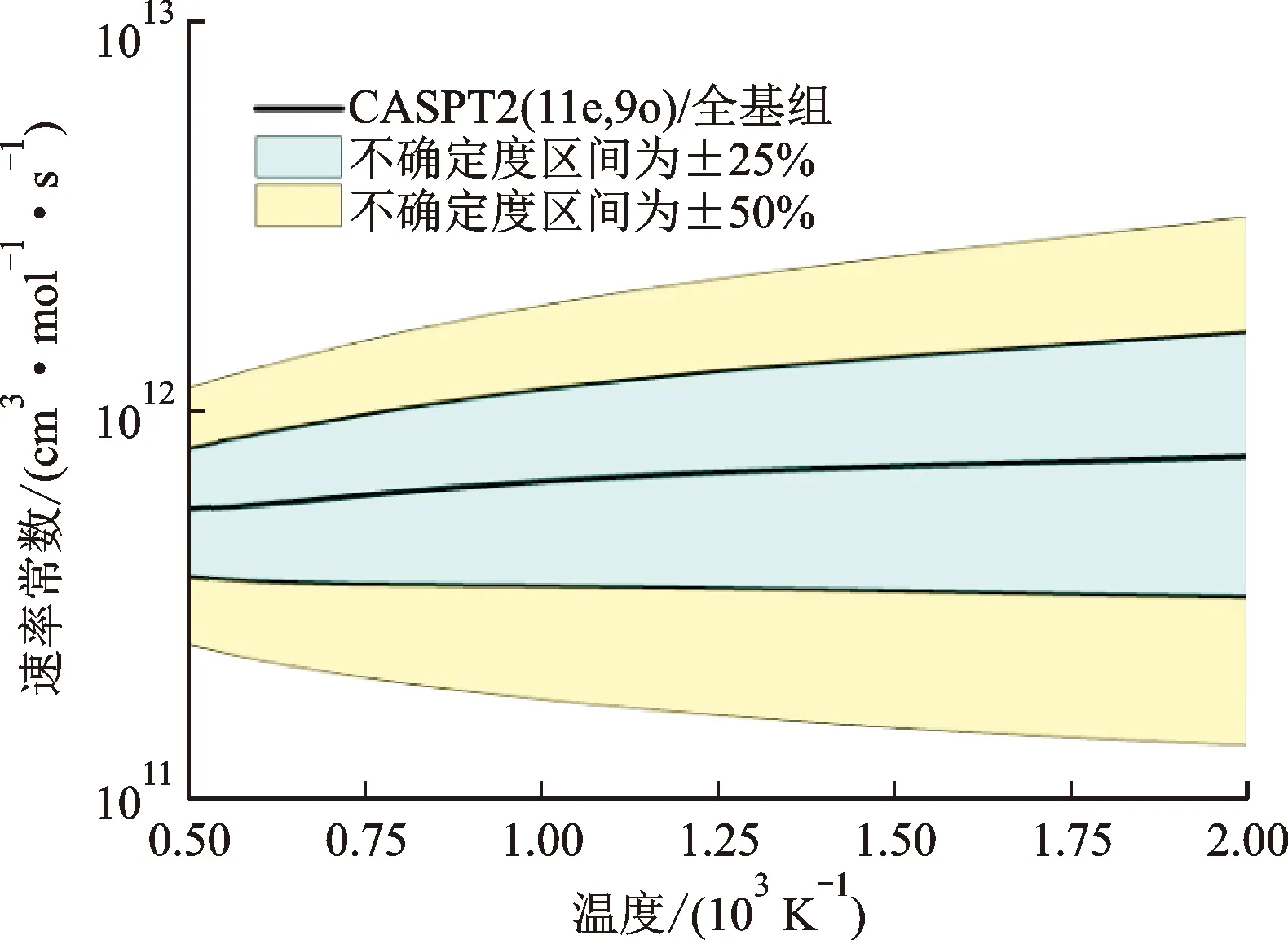
图8 CH2CO(O)H+O2→O2CH2OOH反应速率常数的不确定度区间
气相单分子反应理论(RRKM)和主方程理论求解的误差来源主要为势垒、频率和碰撞参数,采用蒙特卡罗取样得到了反应速率常数的不确定度范围及不确定度如图9、图10所示。CH2CO(O)H+O2→CH2OOH+CO2的反应速率常数不确定度随着温度升高而降低,在低温下约为1.7,在温度高于1 000 K时约为1.2。该反应的不确定度不随压力发生明显的变化,说明反应对碰撞参数不敏感。CH2CO(O)H分解反应和异构化反应的不确定度范围及不确定度如图12、图13所示。在 300 K时约为2.6,在温度高于1 000 K时约为1.2。
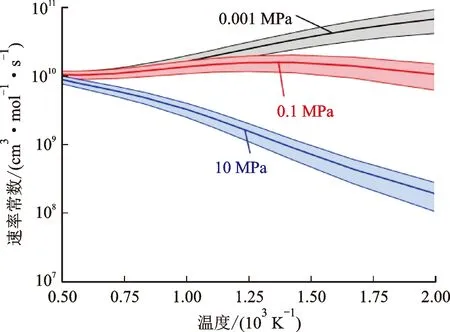
图9 CH2CO(O)H+O2→CH2OOH+CO2反应速率常数的不确定度区间
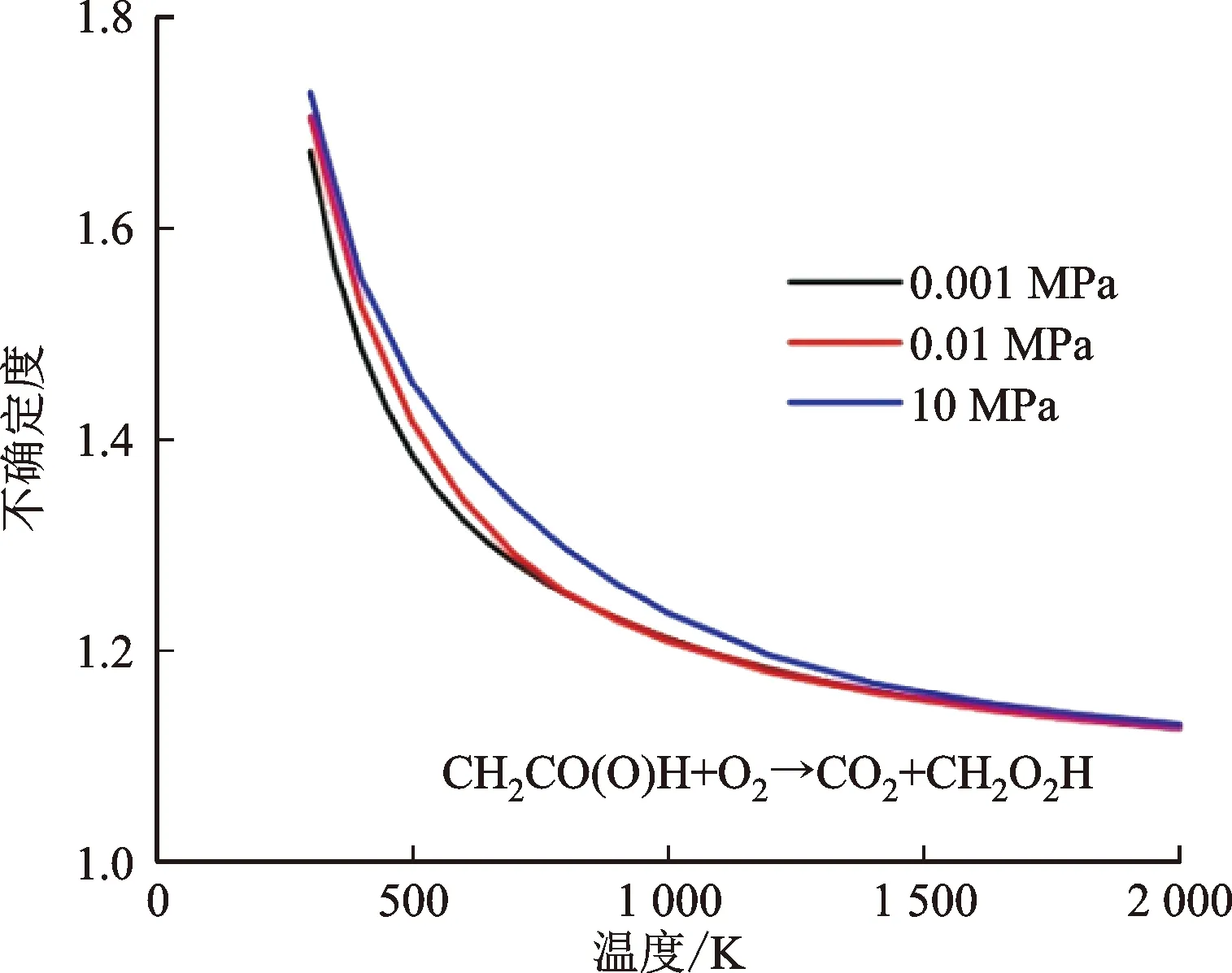
图10 CH2CO(O)H+O2→CH2OOH+CO2的反应不确定度
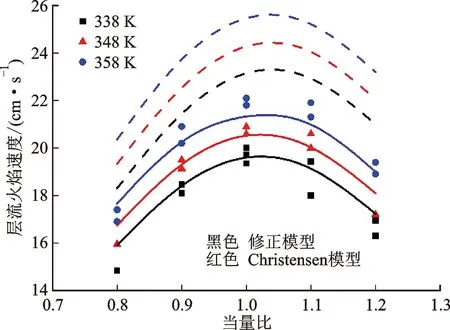
图11 修正模型模拟值、Christensen模型模拟值与实验结果的对比
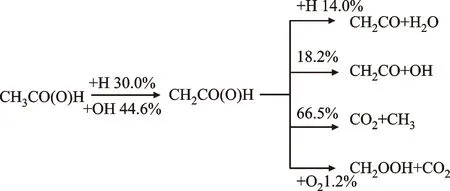
(a)修正模型
2.4 乙酸模型的修正
原有的Christensen模型[14]中缺失了自由基+O2的反应簇,自由基分解生成CO2+CH3,并且OH+CH2CO→CH2CO(O)H的反应速率常数被粗略的估计为4×1012cm3·mol-1·s-1。为了优化模型且研究以上反应体系的竞争关系,将计算的温度压力依赖的速率常数带入到Christensen模型[14]中,填补了其缺失的路径,更新了陈旧的速率常数,模拟了乙酸的层流燃烧速度,与文献中的实验值[11]进行对比,结果如图11所示,实验工况为:p=0.1 MPa,乙酸空气燃烧。修正后的模型能够更好地预测层流燃烧速度。
为了探明修正后模型能够准确预测乙酸的层流火焰速度的原因,本文开展了p=0.1 MPa、T=358 K、当量比φ=1.0条件下的乙酸消耗路径分析。在修正模型中,生成的CH2CO(O)H有4条反应通道,直接分解为CO2+CH3占据主导地位,分支比为66.5%,与CH2CO(O)H分解势能面一致。CH2CO(O)H会与O2加成发生化学活化反应,生成CH2OOH+CO2。该反应路径是通过量化计算后新添加的,证明了量化计算的重要性,可以填补原模型中缺失的路径。由图12(b)可知,在Christensen模型中,乙酸首先发生脱氢反应后生成CH2CO(O)H,97%的CH2CO(O)H自由基直接分解为CH2CO+OH,生成过多OH基促进反应活性,因此模拟的层流火焰速度远高于实验值。
3 结 论
本文利用高精度的量化计算,得到CH2CO(O)H与O2的双分子反应及后续分解反应,CH2CO(O)H异构化和自分解反应的速率常数,包含高压极限和压力相关的速率常数获得结论如下。
(1)在低压或者高温条件下超过99%的化学活化的O2CH2CO2H自由基直接分解为CH2O2H+CO2。随着压力升高化学活化的O2CH2CO2H碰撞稳定后生成O2CH2CO2H,O2CH2CO2H则在后续反应中分解为CH2O2H+CO2。乙酸自由基在温度低800 K时,CO2+CH3产物通道占据主导地位,随着温度升高,CH2CO+OH的通道开始变得更加重要,可以与CO2+CH3反应通道竞争。
(2)反应CH2CO(O)H+O2→CH2O2H+CO2的速率常数不确定度随着温度升高而降低。在低温下约为1.7,在温度高于1 000 K时约为1.2。CH2CO(O)H分解反应和异构化反应的不确定度范围及不确定度在300 K时约为2.6,在温度高于1 000 K时约为1.2。
(3)将计算的温度压力依赖的速率常数带入到Christensen模型中,填补了其缺失的路径,更新了陈旧的速率常数,能够更好地预测层流燃烧速度。在修正模型中,生成的CH2CO(O)H有4条反应通道,直接分解为CO2+CH3占据主导地位,同时会与O2加成发生化学活化反应,生成CH2OOH+CO2。该反应路径是通过量化计算后新添加的,证明了量化计算的重要性。
