牙龈卟啉单胞菌通过CXCL2/CXCR2轴促进口腔鳞癌进展的动物模型研究
2023-06-15热孜万姑丽亚森买热拍提买明李晨曦龚忠诚
热孜万姑丽·亚森, 买热拍提·买明, 李晨曦,3, 龚忠诚
(1新疆医科大学第一附属医院(附属口腔医院)颌面肿瘤外科, 2新疆维吾尔自治区口腔医学研究所, 乌鲁木齐 830054;3口腔颌面发育与再生湖北省重点实验室, 武汉 430022)
口腔鳞状细胞癌(Oral squamous cell carcinoma,OSCC)是头颈部最常见的恶性肿瘤之一,具有高的异质性、复发率、转移率及侵袭率[1]。2020年,全球范围新增OSCC病例377 713例,其中死亡病例177 757例,经过标准化治疗后,患者5年生存率约为50%,复发率高达18%~76%[2]。牙龈卟啉单胞菌(Porphyromonasgingivalis,P.gingivalis)是一种革兰氏阴性专性厌氧菌,具有很强的黏附性,与牙周炎关系最为密切[3]。大量研究表明慢性炎症与癌症的关系,如幽门螺旋杆菌感染与胃癌[4],人乳头瘤病毒感染与宫颈癌[5],EB病毒感染与鼻咽癌[6],乙型肝炎病毒感染与肝癌[7]以及牙龈卟啉单胞菌感染与口腔癌[8]。在OSCC的进展过程中存在多种趋化因子及其受体,主要是CXC类趋化因子,如CXCL1、CXCL2、CXCL5等。研究表明趋化因子配体2/趋化因子受体2(CXCL2/CXCR2)作为潜在致癌因子与多种肿瘤的恶性生物学行为相关[9]。研究发现多种G蛋白在不同癌症(结肠癌、膀胱癌和甲状腺癌)中具有致癌作用,包括CXCL2/CXCR2信号轴的下游蛋白Gαi、Gαq、Gαs,其中最典型的是Gαi[10],故本研究用Gαi的免疫组化检测来验证CXCL2/CXCR2信号轴抑制剂SCH527123是否成功抑制该信号轴。P.gingivalis诱导免疫细胞产生促肿瘤的趋化因子,这些因子与肿瘤细胞的增殖、凋亡、上皮间质转化、侵袭、转移等相关[8]。P.gingivalis感染的OSCC肿瘤微环境中,CXCL2通过肿瘤相关中性粒细胞(Tumor-associated neutrophils,TANs)表面黏附分子CD66b和CD11b趋化TANs向促进肿瘤的N2表型分化, 从而起到促进肿瘤进展的作用[11],故本研究用CD66b来标记N2型TANs。P.gingivalis及CXCL2在OSCC发生发展过程中的作用机制尚未完全明确。因此,在课题组前期研究的基础上通过动物实验来进一步探讨P.gingivalis如何通过CXCL2/CXCR2轴促进口腔鳞癌的进展。
1 材料与方法
1.1 主要试剂P.gingivalis(北纳生物,中国);SCC7鼠源性鳞状细胞癌细胞株(上海富衡生物科技有限公司,中国);CXCL2/CXCR2信号轴抑制剂SCH527123(MCE公司,中国);P.gingivalis一抗(武汉戴安生物公司,中国);Anti-CXCL2、E钙黏蛋白(E-cadherin,E-Cad)、N钙黏蛋白(N-cadherin,N-Cad)一抗,二抗(北京博奥森公司,中国);CD66b+肿瘤相关中性粒细胞抗体ab197678一抗,(Abcam公司,英国);二氨基联苯胺四盐酸盐(diaminobenzidine tetrahydrochloride,DAB)显色试剂盒(Abcam公司,英国);胎牛血清(Gbico公司,美国);小鼠CXCL2 Elisa试剂盒(江莱生物公司,中国);RPMI-1640培养基(Lonza 公司,美国);SYTO9 核酸染料、DAPI 细胞核染料、Mito Tracker Red细胞线粒体染料(懋康生物,中国)。实验动物:C57BL/6小鼠(新疆医科大学动物实验中心,伦理审批号:IACUC20180411-13)。
1.2 方法
1.2.1 SCC7细胞的培养 将冻存的1 mL细胞悬液放在37℃水浴中迅速摇晃解冻,加入5 mL培养基混合均匀。在1 000 r/min条件下离心5 min,弃去上清液,补加4~6 mL完全培养基后吹匀,将所有细胞悬液加入培养瓶中,恒温培养箱(5%CO2,37℃)内过夜,第2天换液并检查细胞密度及贴壁情况。细胞密度达80%~90%时进行传代培养。
1.2.2P.gingivalis的培养 本实验用的P.gingivalis菌株为ATCC33277标准菌株[11](本课题组前期研究已测序验证),用哥伦比亚液体培养基和琼脂固体培养基培养。超净工作台内将细菌接种至哥伦比亚血琼脂平板,将平板和厌氧产气袋放入厌氧箱内,置于37℃恒温箱培养。
1.2.3 SCC7与P.gingivalis的共培养及验证细菌计数 吸取1 mL菌液至含9 mL的哥伦比亚液体培养基的离心管中,混匀,37℃恒温摇床摇匀至生长对数期。酶标仪的OD=450 nm吸光值至1时,细菌浓度为1×109CFU/mL。细菌/细胞的共培养:取生长对数期的SCC7细胞,胰蛋白酶消化后,使用含10%胎牛血清的完全培养基配成细胞悬液。25 mL的培养瓶中接种5×106个细胞后加约5 mL完全培养基。将培养瓶置于细胞培养箱(5%CO2,37℃)中培养8 h,细胞贴壁后弃原培养基,加含2%胎牛血清的低浓度培养基进行培养12 h。感染复数(Multiply of infection,MOI)=50建立共培养模型[12],未加菌液作为阴性对照。激光共聚焦显微镜验证共培养模型,用SYTO9绿色荧光标记P.gingivalis,DAPI蓝色荧光标记细胞核,Mito Tracker Red红色荧光标记线粒体。
1.2.4 C57BL/6小鼠荷瘤实验 用C57BL/6小鼠建立荷瘤模型,用不同浓度CXCL2/CXCR2轴抑制剂干预,将40只雌性C57BL/6小鼠分为对照组(SCC7)、高剂量组(SCC7+P.gingivalis+0.20 nmol/LSCH527123)、低剂量组(SCC7+P.gingivalis+0.05 nmol/L SCH527123)和实验组(SCC7+P.gingivalis)。细胞浓度为2×105个/皿,菌液浓度为1×109CFU/mL,以MOI=50建立共培养模型,腹腔麻醉后于右侧颊黏膜处注射细菌细胞混合悬液200 μL[13],对照组只注射细胞悬液。高、低剂量组于第 4、8、12、16天于右侧颊黏膜造模处分别注射高、低剂量的抑制剂100 μL[14]。于第10、20天两个时间点处死小鼠,切取肿瘤组织后常规石蜡包埋,收取小鼠血液标本。用游标卡尺测瘤体的长径a(mm)、短径b(mm),计算瘤体体积V(mm3)。计算公式[13]为:V=1/2×a×b2。
1.2.5 小鼠肿瘤标本免疫组化实验 常规脱蜡,抗原修复后,一抗在4℃下孵育过夜,一抗的稀释比分别是P.gingivalis(1∶200)、CD66b+(1∶500)、CXCL2(1∶500)、E-Cad(1∶400)、N-Cad(1∶400)、Gαi(1∶200)[14],次日加辣根过氧化物酶标记二抗,DAB显色,苏木素复染,脱水,封片。光镜下观察SCC7组织染色情况,在200倍镜下随机选5个无重复的视野,用Image-Pro plus 6.0软件进行光密度值分析,测量每张图片的累积光密度值(Integrated optical denisty,IOD),阳性区域面积(Area)。结果分级标准:计算出每张图片的平均光密度值,计算公式:平均光密度值=IOD/Area,根据平均光密度值分为阴性(-,0~0.10),弱阳性(+,0.11~0.15),阳性(++,0.16~0.20),强阳性(+++,0.21~0.30)。
1.2.6 Elisa实验 收集小鼠血清用Elisa检测CXCL2含量,将收集于血清分离管的全血标本4℃过夜,1 000 r/min条件下离心20 min,取上清液,按照Elisa试剂盒操作步骤,加样,37℃温育1 h加生物素化抗体:取出酶标板,弃去液体,每孔加入生物素化工作液100 μL,37℃温育1 h。洗板、加酶结合物工作液:洗板3次后每孔加工作液,37℃温育30 min。洗板、加底物:洗板5次后,每孔加入90 μL底物,37℃避光温育15 min。加终止液:每孔加入50 μL,立即在450 nm波长处测定各孔的OD值。
1.3 统计学方法用SPSS 27.0软件进行数据统计学分析,计量资料以均数±标准差表示,组间比较用单因素方差分析和t检验,多个均数的两两比较用SNK法,P<0.05认为差异具有统计学意义。免疫组化的实验结果用Image-Pro plus 6.0 进行光密度值分析。用Graphad Prism 8.0软件进行图像制作。
2 结果
2.1 SCC7与P.gingivalis共培养模型的验证激光共聚焦显微镜图显示,P.gingivalis为绿色荧光,细胞核为蓝色荧光,线粒体为红色荧光,P.gingivalis与SCC7共培养后可见绿色荧光标记的P.gingivalis进入SCC7细胞质中,见图1。
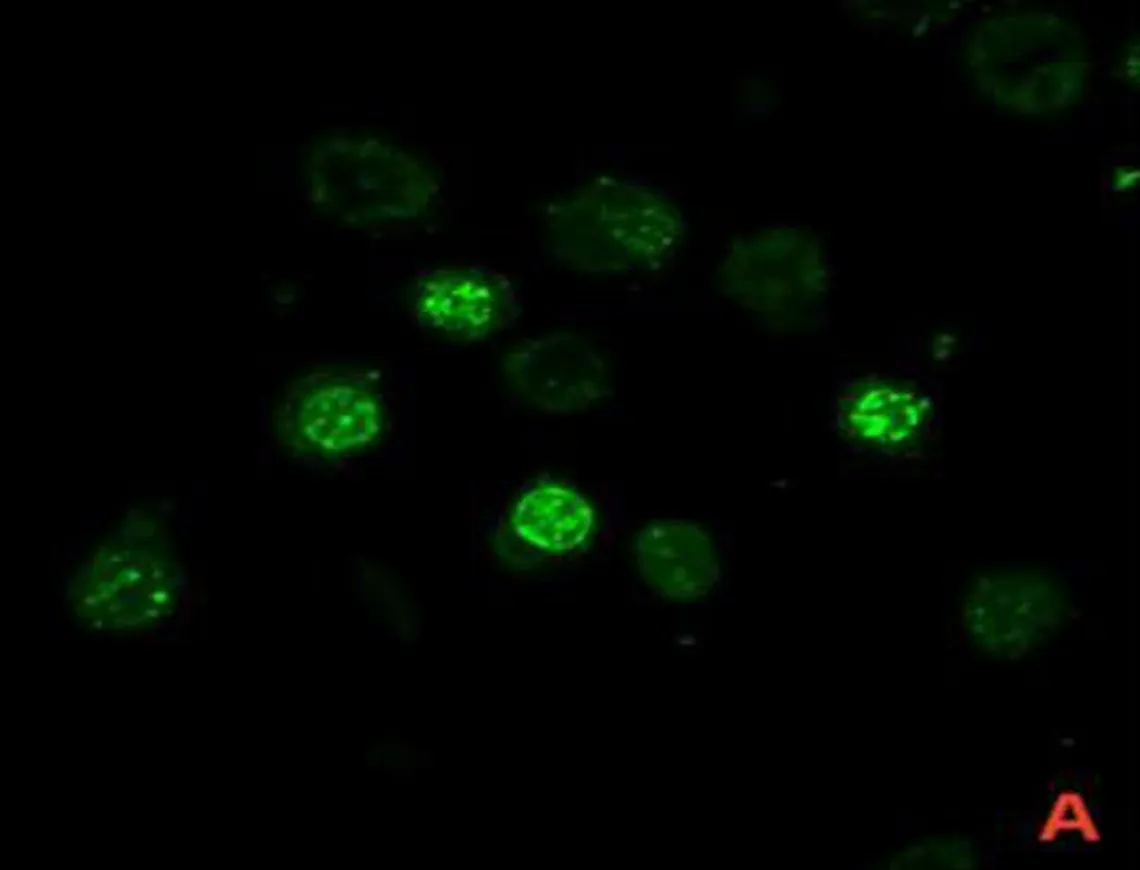
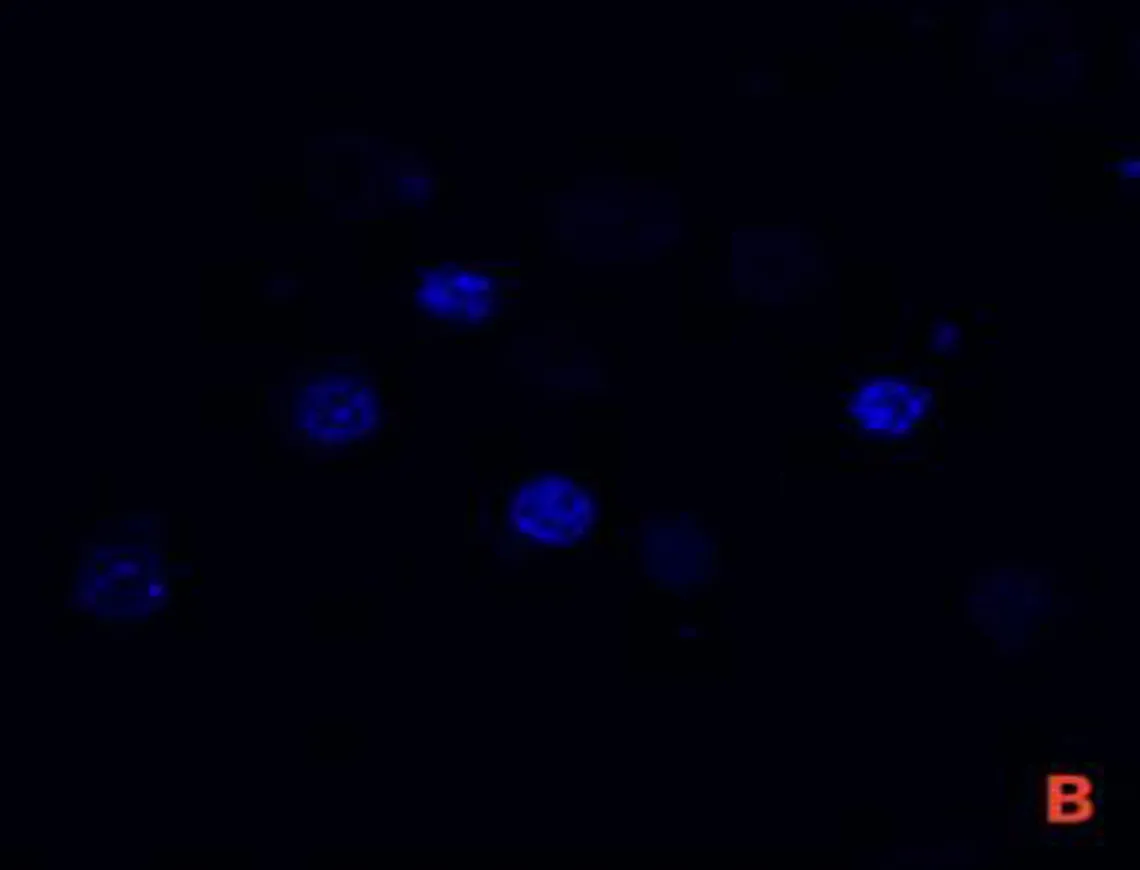

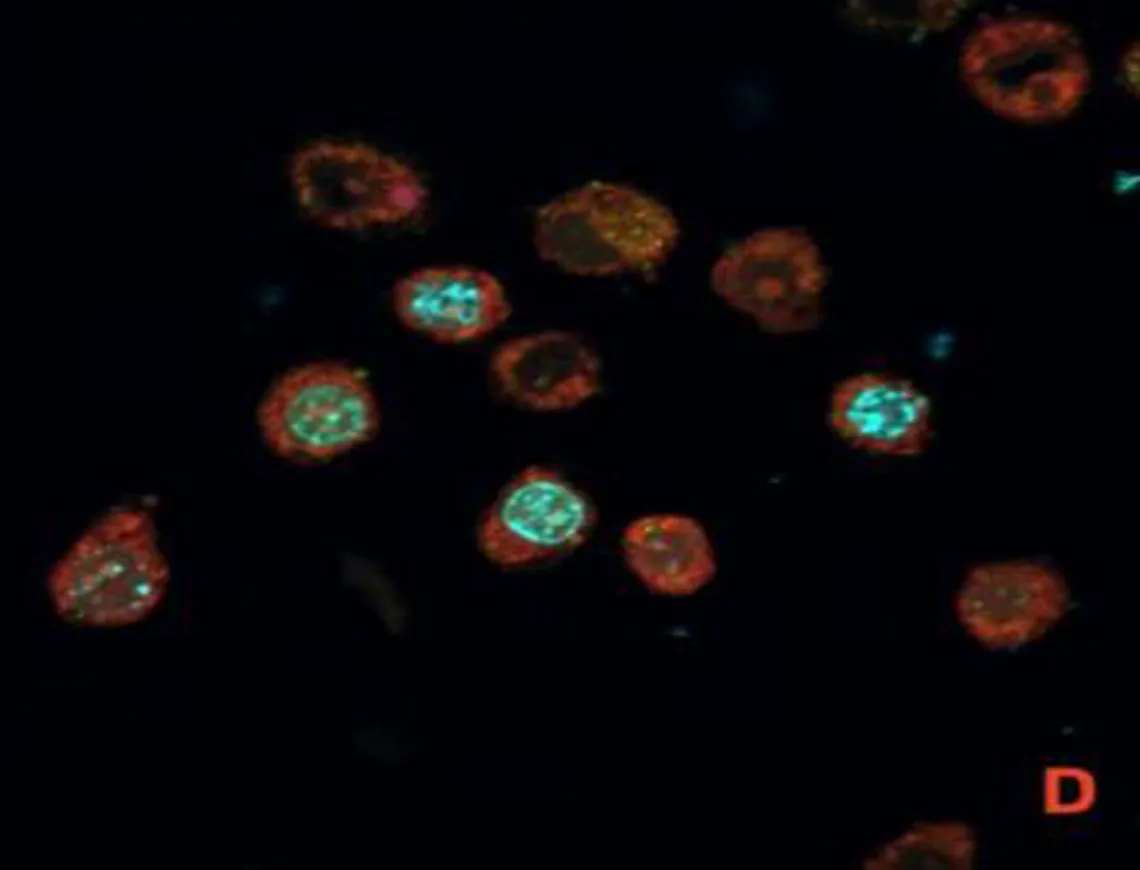
注: A, 被绿色荧光标记的P.gingivalis; B,被蓝色荧光标记的细胞核; C, 被红色荧光标记的线粒体; D, P.gingivalis进入SCC7细胞质中。
2.2 C57BL/6小鼠荷瘤实验各组小鼠分别在第10天、第20天两个时间点处死,完整切取肿瘤组织。同一个组中,第10天与第20天的瘤体体积相比,随着时间的延长瘤体体积增大,对照组、低剂量组、实验组第20天的瘤体体积均大于第10天的,差异有统计学意义(P<0.05),高剂量组第10天与第20天的瘤体体积相比差异无统计学意义(P>0.05)。与对照组相比,低剂量组、实验组的瘤体体积明显增大,差异有统计学意义(P<0.05),对照组与高剂量组瘤体体积相比差异无统计学意义(P>0.05)。用不同剂量的CXCL2/CXCR2轴抑制剂干预时,低剂量组与实验组相比瘤体体积缩小(P<0.05);高剂量组与实验组相比瘤体体积明显缩小(P<0.01),肿瘤抑制更明显,见图2。
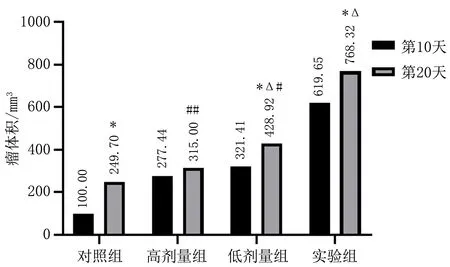
注:与第10天比较, *P<0.05;与对照组比较, △P<0.05; 与实验组比较, #P<0.05, ##P<0.01。
2.3 小鼠肿瘤组织免疫组化实验结果Gαi在对照组和高剂量组中弱阳性表达,低剂量组中阳性表达,实验组中强阳性表达,实验组中的表达强于对照组、高剂量组和低剂量组(P<0.05),低剂量组与高剂量组相比差异有统计学意义,低剂量组中的表达强于高剂量组(P<0.05),表明CXCL2/CXCR2信号轴抑制剂SCH527123成功抑制该信号轴(图3、4)。
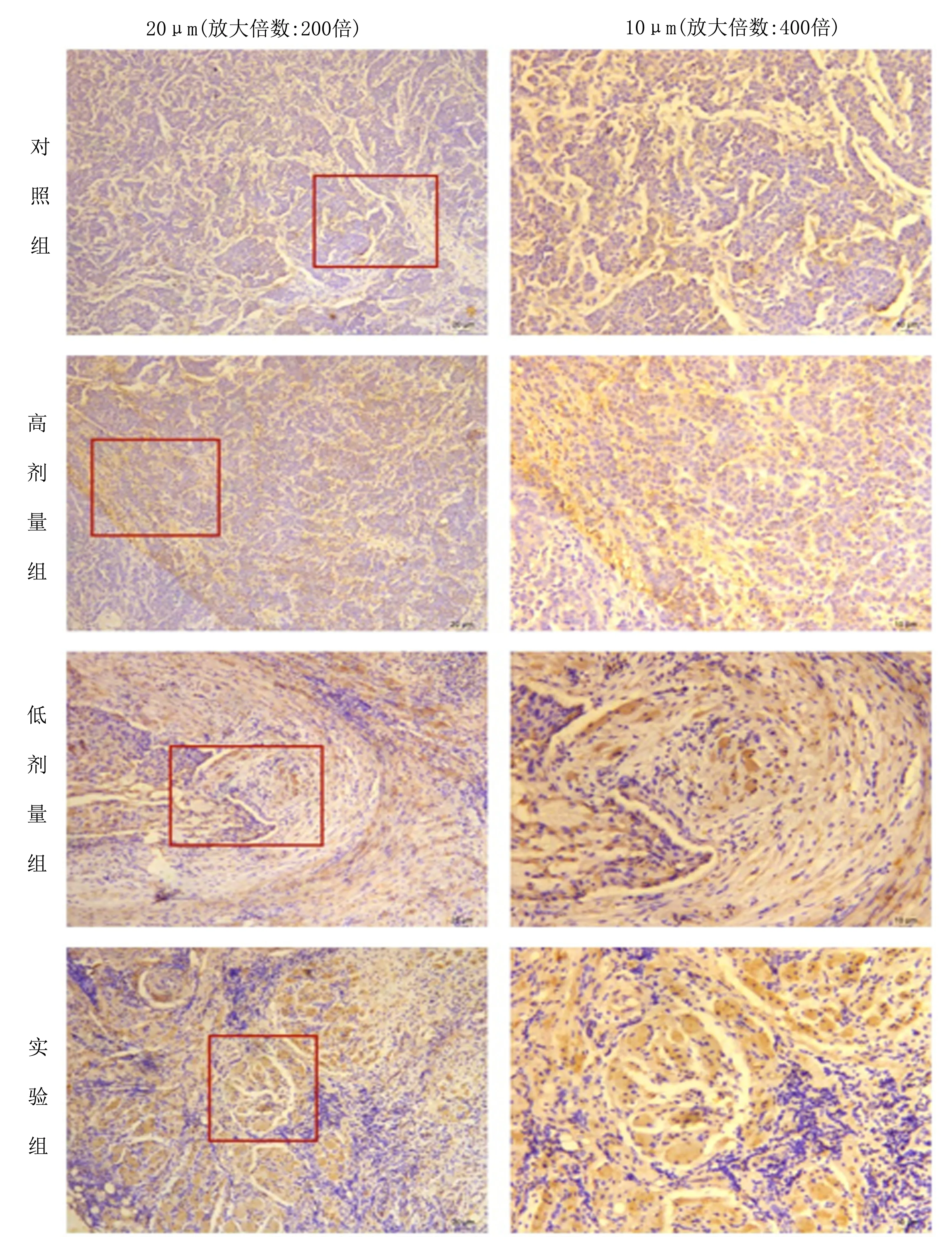
注:左图方框中的区域40×10倍镜下观察后是右图。
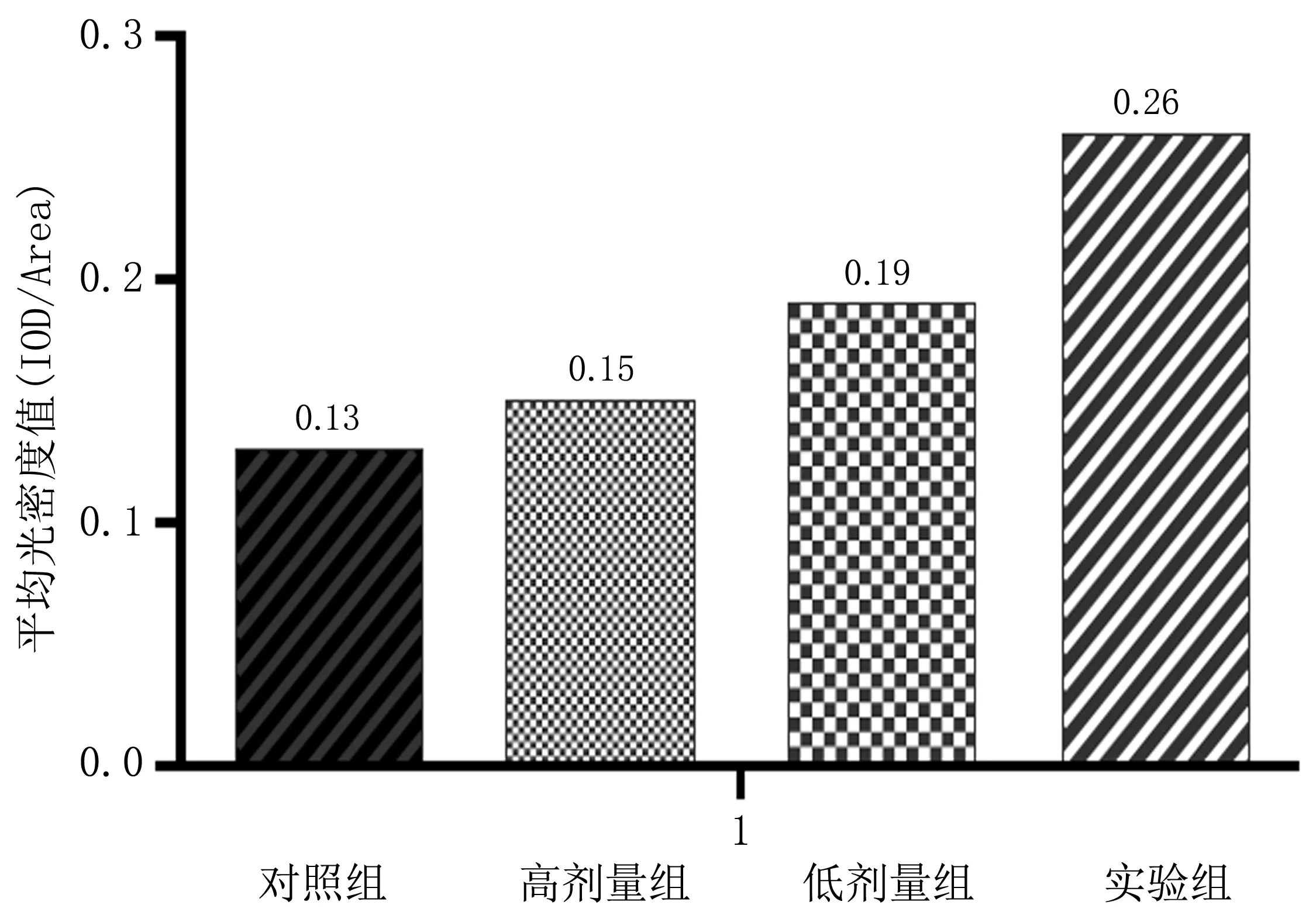
图4 Gαi在各组小鼠肿瘤组织中表达的平均光密度值
2.4P.gingivalis、CXCL2、CD66b+、E-cad、N-cad在各组小鼠肿瘤组织中的表达P.gingivalis在高剂量组和低剂量组中弱阳性表达,在实验组中阳性表达,实验组中的表达强于高剂量组和低剂量组(P<0.05)。CXCL2在对照组中阴性表达,高剂量组中弱阳性表达,低剂量组中阳性表达,实验组中强阳性表达,CD66b+在对照组和高剂量组中阴性表达,低剂量组中弱阳性表达,实验组中阳性表达,CD66b+、CXCL2在实验组中的表达均强于对照组、高剂量组和低剂量组(P<0.05),低剂量组中的表达强于高剂量组(P<0.05)。N-Cad在对照组和高剂量组中弱阳性表达,低剂量组中阳性表达,实验组中强阳性表达,实验组中的表达强于对照组、高剂量组和低剂量组(P<0.05)。E-Cad在对照组中强阳性表达,高剂量组中阳性表达,低剂量组中弱阳性表达,实验组中阴性表达,对照组中的表达强于实验组、高剂量组和低剂量组(P<0.05)。P.gingivalis、CXCL2、CD66b+、N-Cad在各组中的表达趋势基本一致,E-Cad的表达与之相反,见图5、6。
2.5 各组小鼠血清CXCL2含量比较对照组、高剂量组、低剂量组和实验组小鼠血清中CXCL2的含量分别为(38.11±1.06)、(62.83±1.17)、(175.68±3.88)和(612.93±1.14)pg/mL,各组间差异均有统计学意义(F=95.955,P=0.000)。实验组CXCL2的含量与对照组、高剂量组、低剂量组相比差异均有统计学意义(P<0.001);低剂量组与高剂量组相比差异有统计学意义,低剂量组CXCL2的含量明显高于高剂量组(P<0.001)。
3 讨论
OSCC作为口腔颌面部最常见的恶性肿瘤,常发生于唇、口腔前庭、牙龈、磨牙后区、口底、颊黏膜、舌、腭等部位。口腔内定植多种细菌,细菌生态系统之间以及细菌与宿主之间存在着复杂的动态平衡,这种平衡失调时微生物尤其是P.gingivalis等优势菌通过释放毒素、代谢产物来刺激局部和全身炎症,改变宿主的免疫反应来促进OSCC的发生发展[15]。研究表明,P.gingivalis能够长时间与发生于口腔内的肿瘤接触,影响肿瘤细胞的上皮间质转化、增殖、凋亡、转移侵袭[8]。
TANs有N1、N2两种极化的表型,N1表型抑制肿瘤,N2表型促进肿瘤。在肿瘤发生发展的不同阶段TANs有不同的表型,早期主要表现为N1表型,随肿瘤的不断进展转型为N2表型并促进肿瘤细胞进入血管,为肿瘤细胞的生存和增殖提供有利条件,进一步促进肿瘤的侵袭转移[16]。相关研究报道,P.gingivalis能够招募TANs介导OSCC的进展[17]。本研究用CD66b+标记N2型TANs,免疫组化结果显示实验组中阳性表达,对照组中阴性表达,并与P.gingivalis的表达趋势一致,这一结果进一步说明P.gingivalis与TANs趋化能力的相关性。
趋化因子是一种小分子分泌型蛋白,以高亲和力与其受体结合,产生“受体-配体”相互作用及强烈的信号级联反应,在肿瘤细胞的活化、分化等恶性生物学行为中起重要的作用[18]。研究表明[9],CXCL2/CXCR2信号轴与多种肿瘤的侵袭转移等恶性生物学行为相关。CXCL2是CXC类趋化因子的成员,与其受体CXCR2结合产生促进肿瘤血管形成的趋化因子[18]。相关文献报道,P.gingivalis能够活化免疫细胞产生具有促进肿瘤恶性生物学行为的趋化因子(CXCL1、CXCL2、CXCL5等)[8]。本研究结果显示小鼠血清中CXCL2的含量以实验组中最高,其次为低剂量组,低剂量组CXCL2的含量明显高于高剂量组。同样免疫组化结果示,CXCL2和P.gingivalis在各组中的表达趋势一致;荷瘤实验进一步验证P.gingivalis通过上调CXCL2的含量来促进肿瘤的进展,使用CXCL2/CXCR2信号轴抑制剂时肿瘤生长被抑制,瘤体缩小,高剂量组更为明显。因此本研究在组织水平和体外实验观察到了相符的结论。
上皮间质转化(Epithelial mesenchymal transition,EMT)是上皮细胞失去极性及细胞间的连接,转化为间质细胞从而获得迁移侵袭能力的过程[19]。研究证实,P.gingivalis在OSCC的进展过程中促进口腔上皮细胞向间质细胞转化,使口腔上皮细胞丧失黏附能力,从而在肿瘤细胞的侵袭转移,增殖等恶性细胞行为中起重要的促肿瘤作用[20]。本研究用具有典型上皮特性的E-cad和典型间质特性的N-cad来标记肿瘤进展中的EMT现象。免疫组化结果示N-Cad在各组中的表达趋势与P.gingivalis、CXCL2、CD66b+的表达趋势一致,而与E-Cad的表达趋势相反,我们推测P.gingivalis通过CXCL2/CXCR2信号轴促进肿瘤EMT现象。
综上所述,本研究通过口腔癌动物模型进一步研究了P.gingivalis、CXCL2、TANs、E-cad、N-cad在OSCC组织中的表达水平及相关性以及CXCL2/CXCR2轴抑制剂对肿瘤的抑制作用,推测P.gingivalis与CXCL2之间可能有直接或间接的作用关系,P.gingivalis可能通过CXCL2/CXCR2轴调控肿瘤细胞的增殖、凋亡、上皮间质转化、侵袭转移等恶性细胞学行为,进一步促进OSCC的进展。目前P.gingivalis促进OSCC的机制尚未完全明确,因此P.gingivalis对OSCC及其TME中细胞的调控机制需要进一步深入研究。
