基于高通量测序分析高粱紫斑病叶片的微生物组成及多样性
2023-06-14罗文梅杨远桥孙静娥王勇任明见
罗文梅 杨远桥 孙静娥 王勇 任明见


摘 要: 本研究采用ITS高通量测序(扩增子测序)技术对已感染紫斑病(A和B)和未感染紫斑病(CK)的高粱叶片进行检测,以明确其真菌的群落组成及多样性。结果表明:高粱紫斑病病叶中真菌菌群组成丰富,总共检出820种真菌,3个样品共同拥有的为155种,所有菌群分属于9门29纲60目122科和206属。3份样品Shannon指数分别为3.98、4.67、4.11,按大小排序为B>CK>A。同时,含量排名靠前的属之间亲缘关系比较复杂,且没有出现前人报道的高粱尾孢 Cercospora sorghi ,可能的原因是引起该病害发生的病原菌发生了变化,或是存在由多种真菌共同作用从而导致病害发生的可能性。本研究结果能帮助我们更好地了解高粱紫斑病发病期叶片真菌的组成和分布的多样性,为该病害的研究和防治提供更好的指导作用。
关键词: 高通量测序;高粱紫斑病;ITS;分类
中图分类号:S432
文献标识码:A
文章编号:1008-0457(2023)02-0017-06
国际DOI编码:10.15958/j.cnki.sdnyswxb.2023.02.003
高粱是禾本科一年生草本植物,具有许多优质的特点,例如抗旱、耐涝、耐贫瘠等,是世界上重要的禾谷类作物之一,据不完全统计,其种植总面积在世界排名前五[1]。高粱用途广泛,可用于酿酒、加工成生物燃料或建筑材料、喂食家养牲畜等[2-4]。同时,高粱不仅在国内备受欢迎,在西方国家也逐步广泛出现,这进一步表明该产业有着巨大的潜力[5]。然而随着高粱种植面积的扩增,其病虫害的发生日益严重。据报道,病害发生程度一般时高粱的产量损失在10%~20%,严重时可达到70%[6]。据报道,我国高粱病虫害大约有100多种,其中真菌病害有28种[7],例如造成叶部危害的病害有炭疽病、锈病、紫斑病、大斑病和纹枯病等[8];造成穗部危害的病害有黑穗病[9];造成茎部病害的有茎腐病[10]。在国外,高粱病害的发生也十分严重,Nutsugah等[11]通过对高粱的病害流行调查发现上述病害普遍存在。
仁怀市是贵州省遵义市一个县级市,位于贵州西北部、赤水河中游,夏季特有的高温多雨以及地理环境,十分适宜高粱的生长,因而成为了茅台酒的故乡[12]。但高温高湿的环境同样也增加了高粱病虫害的发生,其中紫斑病是高粱常见的真菌病害之一,严重影响了其产量和品质[13-14]。高通量测序技术的发展不仅提升了我们对微生物种群的组成了解,也帮助我们能更好地分析微生物菌群的多样性[15-18]。同时,已有研究表明基于高通量测序技术可以帮助真菌病害病原菌的鉴定,如通过该技术帮助鉴定红枣黑斑病[19]和草莓白粉病的病原菌[20]。但是,至今尚未有具体的报道对高粱紫斑病发生时微生物种群的组成情况进行分析,基于此本研究通过高通量测序技术对发病时期高粱紫斑病菌的菌群群落结构差异比较分析,以期有利于进一步分析高粱紫斑病的发病机制,为高粱紫斑病的防治提供更好的理论依据。
1 材料与方法
1.1 试验材料
供试材料为高粱紫斑病病叶,于2021年8月采集自贵州省遵义市仁怀市。共3份样品,A(症状较轻)、B为感病叶片,CK为健康叶片。
1.2 基因组DNA的提取和PCR扩增
采用CTAB方法对样本的基因組DNA进行提取,利用琼脂糖凝胶电泳检测DNA的纯度和浓度,以稀释后的基因组DNA为模板,根据测序区域的选择,使用带Barcode的特异引物,New England Biolabs公司的Phusion High-Fidelity PCR Master Mix with GC Buffer和高效高保真酶进行PCR,确保扩增效率和准确性。引物为ITS1(ITS5-1737F和ITS2-2043R)。
1.3 PCR产物的混样和纯化
PCR产物使用2%浓度的琼脂糖凝胶进行电泳检测;对检测合格的PCR产物进行磁珠纯化,采用酶标定量,根据PCR产物浓度进行等量混样,充分混匀后使用2%的琼脂糖凝胶电泳检测PCR产物,对目的条带使用qiagen公司提供的胶回收试剂盒回收产物。
1.4 文库构建和上机测序
使用TruSeq DNA PCR-Free Sample Preparation Kit建库试剂盒进行文库构建,构建好的文库经过Qubit和Q-PCR定量,文库合格后,使用NovaSeq6000 进行上机测序。
1.5 信息分析部分
1.5.1 测序数据处理
根据Barcode序列和PCR扩增引物序列从下机数据中拆分出各样本数据,截去Barcode和引物序列后使用FLASH(V1.2.7,http://ccb.jhu.edu/software/FLASH/)对每个样本的reads进行拼接,得到的拼接序列为原始Tags数据(Raw Tags);拼接得到的Raw Tags,经过严格的过滤处理[21]得到高质量的Tags数据(Clean Tags)。
1.5.2 OTU聚类和物种注释
利用Uparse算法对所有样本的全部Effective Tags进行聚类,默认以97%的一致性(Identity)将序列聚类成为OTUs(Operational Taxonomic Units), 同时选取OTUs的代表性序列,依据其算法原则,筛选的是OTUs中出现频数最高的序列作为 OTUs的代表序列。 对OTUs序列进行物种注释,用Mothur方法与SILVA138(http://www.arb-silva.de/)[22]的SSUrRNA数据库进行物种注释分析(设定阈值为0.8~1),获得分类学信息并分别在各个分类水平:kingdom(界),phylum(门),class(纲),order(目),family(科),genus(属)统计各样本的群落组成。
1.5.3 样本多样性分析
使用Qiime软件(Version 1.9.1)计算Observed-otus, Chao1,Shannon,Simpson,ace,Goods-coverage,PD_whole_tree指数。
2 结果与分析
2.1 测序数据处理与OTU分析
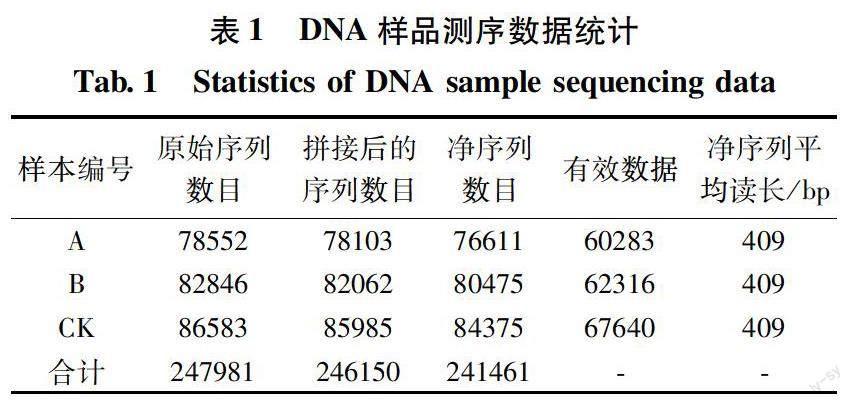
从3份测序样品(样品A,样品B和CK)中共获得247981条原始测序Reads,数据质控(QC)后得到241461条净数据Reads,其平均长度为409 bp(表1)。完成数据质控和去除嵌合体分析后,将拼接序列聚类成物种OTU,总共得到820条物种OTU;A、B、CK分别含有256、302、262条OTU,所有样品共享155条物种OTU(图1)。
2.2 物种分布情况
通过对测序结果进行整理,并在门、纲、目、科、属5级分类水平上水平对各个样品进行分类(表2),总共注释得到9门、29纲、60目、122科和206属。图2和图3表明,样本A与B存在明显差异,但样本A与样本CK的菌群组成比较相似。再由表2可知,感病前后真菌群落组成结构基本相同,差异较大的是不同分类水平下的相对丰度。在门分类水平担子菌门Basidiomycota和子囊菌门Ascomycota 为优势菌群,且感病后Ascomycota在B中的丰度明显大于CK;在纲分类水平座囊菌纲 Dothideomycetes和银耳纲Tremellomycetes为优势菌群,在目分类水平银耳目Tremellales和叶黑粉菌目Entylomatales为优势菌群,在科分类水平Entylomatales和牛肝菌科Bulleribasidiaceae为优势菌群,在属分类水平上,按相对丰度排序靠前的顺序分别为 Tilletiopsis 、枝孢属 Cladosporium 、亚隔孢壳属 Didymella 、汉纳酵母属 Hannaella 、附球菌属 Epicoccum 、 Dioszegia 、 Papiliotrema 、 Apiotrichum 。样本B中 Cladosporium 和 Epicoccum 的相对丰度大于CK,其余属相对丰度在CK中均大于样本B。因此根据相应数据推测,高粱紫斑病害的前后期叶片的真菌群群丰度发生了较大的变化,可能存在多种致病菌共同作用的情况。
2.3 高粱紫斑病病叶菌群的多样性
由表3可知,Goods Coverage均为1.00,说明试验测序结果能够准确的反应供试样品真实情况。在同一时期,3个样本的Chao1指数大小顺序为B>CK>A,ACE指数为269.58~315.52,PD whole tree指数在57.34~71.68,表明本次多样性测序深度达到了丰富度估计值,而且样本中的绝大部分真菌物种信息都能被本研究捕捉到,但不同菌种间亲缘关系差异较大。同时,样本的Shannon指数大小顺序为B>CK>A,Simpson指数大小排序也相同,上述结果均说明同一时期的样本B真菌群落多样性高于CK真菌群落多样性。
3 结论与讨论
随着高通量测序技术的日趋成熟,其应用范围越发广泛。目前,已有运用该技术对高粱根际微生物群落[23]、雪茄烟微生物群落[24]、健康桑果和患病桑果内生菌群落结构[25]等各方面的研究。李祥栋等[26]通过ITS高通量测序技术对薏苡的黑穗病瘿的真菌进行检测,发现了黑穗病害的致病菌为 Sporisorium 与 Ustilago ,其中 Sporisorium 属于优势菌群,该结论与前人报道的薏苡黑穗病菌的鉴定并不相同[27],推测薏苡黑穗病菌存在多种变异类型。有研究表明,同一寄主在不同产地的不同生长时间里的病原菌不一定相同[28],因而对单一的病原菌进行分离和回接并不能准确的认识各个病害的病原菌,这也是高通量测序技术被更广泛用于患病植株微生物群落结构分析的原因之一。对于高粱紫斑病,有报道表明引起该病害的病原菌为 Cercospora sorghi [29-30]。但本研究的基因组测序结果表明,在所采集的紫斑病病叶上微生物组成含量列表排名靠前的属中并没有 Cercospora 。病害样本中A与B中 Cercospora 丰度比例仅分别为2.16%与0.15%,其含量并不占优势。加之在初期的病样分离时并没有分离得到 Cercospora 所属的菌株。因此,不排除在贵州省遵义市仁怀市的高粱紫斑病病害叶片中,其病原菌已经发生了变化。当然也可能由于 Cercospora sp.生长比较慢且分离比较困难,故而我们没有分离到。
本文基于对所采患紫斑病的高粱病叶与健康叶片进行高通量测序,并对其结果进行分析得出:(1)高粱感染紫斑病后,叶片的真菌菌群相对丰度发生了明显变化,与健康叶片菌群相比,在一定程度上打破了原有的微生物生态平衡,形成了新的真菌群落关系;(2)患病样本B中 Cercospora 含量极少,不排除在贵州省遵义市仁怀市的高粱紫斑病病害叶片中,该病害的病原菌已经发生了变化。
综上所述,高粱紫斑病的病原菌也许已开始变得复杂多样,通过高通量测序技术的分析帮助我们更加清晰地了解发病前后其真菌群落的变化情况。因此对该病害病原菌的准确鉴定及发病规律研究将是以后需要全面研究的方向,以期为该病害的防治提供更好的理论指导。
(责任编辑:段麗丽)
参 考 文 献:
[1]
王瑞,王金胜,张福耀,等.1970s—2000s中国高粱杂交种亲本遗传距离演变的SSR分析[J].中国农业科学,2015,48(3):415-425.
[2] 程度,曹建兰,王珂佳,等.高粱对酱香型白酒品质影响的研究进展[J].食品科学,2022,43(7):356-364.
[3] Dahlberg J,Berenji J,Sikora V,et al.Assessing sorghum [Sorghum bicolor(L.) Moench] germplasm for new traits:food,fuels & unique uses[J].Maydica,2011,56(2):85-92.
[4] Dahlberg J.The Role of Sorghum in Renewables and Biofuels[J].Methods in Molecular Biology,2019,1931:269-277.
[5] Rumler R,Bender D,Schnlechner R.Sorghum and its potential for the Western diet[J].Journal of Cereal Science,2022(1):104.
[6] 李祥艳,许文志,林超文,等.西南地区高粱常见病虫害和鸟害的发生与防治[J].四川农业科技,2022(3):56-57,64.
[7] 徐秀德,刘志恒.高粱病虫害原色图鉴[M].北京:中国农业科学技术出版社,2012.
[8] 陈光达.有机高粱常见病虫害防治技术初探[J].南方农业,2017,11(30):13-14,16.
[9] 潘明凤,魏媛.高粱种植基本技术及常见病虫害防治措施[J].农村经济与科技,2021,32(5):74-75.
[10] 黄敏佳.高粱镰孢菌茎腐病研究进展[J].中国农学通报,2016,32(14):90-95.
[11] Nutsugah S K,Atokple I D K,Leth V.Sorghum diseases prevalent in Ghana[J].Ghana Journal of Agricultural Science,2008,40(2):119-126.
[12] 姜培跃.仁怀市高粱主要病虫害综合防治措施[J].植物医生,2016,29(8):57-58.
[13] 苟开礼,李士敏.不同育苗方式对高粱红缨子产量及其紫斑病发生的影响[J].农技服务,2019,36(10):28-29,31.
[14] 董亚.务川县酒用高粱的栽培管理技术[J].江西农业,2020(8):29,32.
[15] 秦楠,栗东芳,杨瑞馥.高通量测序技术及其在微生物学研究中的应用[J].微生物学报,2011,51(4):445-457.
[16] 李凤,董晓明,董志新,等.四川盆地典型城市与农村河流微生物群落特征比较分析[J].山地学报,2022,40(1):56-70.
[17] 林庆柱,林芬,蔡林生,等.基于高通量测序的猕猴桃根腐病根际土壤真菌群落研究[J].落叶果树,2022,54(4):43-46.
[18] 康捷,章淑艳,韩韬,等.两种麻山药典型病害根际土壤微生物多样性的研究[J].生物技术通报,2017,33(7):107-113.
[19] 刘晓勤,张锐利.基于高通量测序技术的红枣黑斑病病原菌分离与鉴定[J].江苏农业科学,2020,48(24):108-112.
[20] 张振荣,田云霞,董琼娥,等.基于高通量测序的草莓白粉病病原菌分析[J].西南农业学报,2021,34(7):1439-1443.
[21] Nicholas A B,Sathish S,Jeremiah J F,et al.Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing[J].Nature Methods,2013,10(1):57-59.
[22] Wang Q.Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy[J].Applied and Environmental Microbiology,2007,73(16):5261-5267.
[23] 刘婷,肖仲久,李小霞,等.高通量测序技术分析茅台酿酒高粱根际真菌群落特征[J].内蒙古农业大学学报(自然科学版),2022,43(1):48-52.
[24] 叶长文,李璐,贺琛,等.基于高通量测序的雪茄烟微生物群落结构和多样性分析[J].烟草科技,2021,54(8):1-9.
[25] 彭芳芳,魏召新,李勋兰,等.基于高通量测序分析感染菌核病和健康桑果内生菌群落结构及多样性[J].食品科学,2021,42(20):61-68.
[26] 李祥栋,潘虹,陆秀娟,等.薏苡黑穗病菌的宏基因组测序及分类解析[J].基因组学与应用生物学,2021,40(1):270-278.
[27] Zhang J Z,Guan P G,Tao G,et al.Ultrastructure and phylogeny of Ustilago coicis [J].Journal of Zhejiang University-Science B(Biomedicine & Biotechnology),2013,14(4):336-345.
[28] 高芬,任小霞,王夢亮,等.中草药根腐病及其微生物防治研究进展[J].中国中药杂志,2015,40(21):4122-4126.
[29] 王春雷.黑龍江省高粱主要病害的发生及防治[J].现代农业科技,2018,(17):119,123.
[30] Jiang Y,Xu J,Hu L,et al.First report of sorghum zonate leaf spot caused by Gloeocercospora sorghi in China[J].Plant Disease,2018,102(5):1033.
Analysis of Microbial Composition and Diversity of Sorghum Purple Spot Leaves based on High-throughput Sequencing
Luo Wenmei1,Yang Yuanqiao1,Sun Jinge1,Wang Yong1*,Ren Mingjian2
(1.Key Laboratory of Agricultural Microbiology,College of Agriculture,Guizhou University,Guiyang,Guizhou 550025,China; 2.College of Agriculture,Guizhou University/ Guizhou Subcenter of National Wheat Improvement Center,Guiyang,Guizhou 550025,China)
Abstract:
ITS high-throughput sequencing technology was used to detect sorghum leaves infected and uninfested with purple spot disease,to determine the composition and diversity of fungal community.The results showed that a total of 820 species OTUs were obtained from fungal community in sorghum purple leaf spot,155 OTU species were shared by the three samples.All these species belonged to 9 phylum,29 classes,60 orders,122 families and 206 genus.The Shannon index relation of the three samples was 3.98,4.67 and 4.11,respectively,which were in the order of B>CK>A.Genetic relationship among the top ten genus were relatively complex,and Cercospora sorghi reported by previous reports was not included in them.We presumed that the pathogen changed or was caused by the combined action of multiple fungi.The results of this study could help us better understand the composition and distribution diversity of leaf fungi during the onset of sorghum purple spot disease.It has a better guiding function for the research and control of this disease.
Keywords:
high-throughput sequencing; purple spot disease; ITS; taxonomic
