橙色红曲菌Ku70基因缺失菌株的构建及功能分析
2023-06-07焦雪雪李燕萍
焦雪雪,李燕萍*
(南昌大学a.食品科学与技术国家重点实验室;b.中德联合研究院;c.食品学院,江西 南昌 330047)
红曲菌是一种重要的传统药食同源菌,能够产生多种对人体有益的代谢产物,其以粳米为基质培养得到的红曲米具有除湿痰、化血化瘀和健脾消食等药用功能[1-3]。红曲菌的有益代谢产物Monacolin K是一种高效降脂产品[4],代谢产物色素是一种应用广泛的天然食用色素,可作为多种慢性疾病甚至癌症的潜在治疗物[5],所以红曲菌是一种在医疗工业中具有重要意义的商业生物。但是红曲菌产生的有益代谢产物产量偏低,且能产生具有肾毒性、肝毒性和致癌活性的桔霉素,严重限制了红曲菌的应用[6-8]。近年来,多项研究涉及在提高红曲菌有益代谢产物的同时抑制甚至消除桔霉素的产生,通常采用三种策略来实现。第一种是优化生理生化过程,包括培养基选择、培养参数(pH值、溶氧量、时间)和发酵模式(补料分批或连续模式)[2,9-11]。第二种策略是突变选育,利用物理、化学等因素诱发生物产生突变,然后通过大量的筛选工作来获得桔霉素含量降低的菌株[12]。第三种策略是通过基因编辑技术,构建基因敲除菌株或异源表达菌株来研究生物体某一段基因的生理功能,推断出其代谢途径,进而引导其代谢流向有益方向[13-15]。
基于同源重组的基因敲除工程菌的构建是研究红曲菌代谢途径和改造红曲菌株的有效方法。该方法的转化效率与生物体内非同源末端连接(non-homologous end joining,NHEJ)和同源重组修复(homo-logous recombination,HR)两种DNA双链断裂修复机制有关[16]。但由于真菌中NHEJ相对于HR具有极大优势,导致真菌中基因靶向效率低。在研究丝状真菌某基因簇功能时,通常需要敲除多个基因,通过大量的筛选工作才能得到阳性敲除菌株[17]。由于生物体内HR和NHEJ存在相互竞争的关系,故可以通过抑制NHEJ途径而提高HR发生概率[18]。真核生物中的NHEJ系统与DNA依赖蛋白激酶密切相关,Ku70/80异源二聚体是DNA依赖蛋白激酶的主要组成部分[19]。其中Ku70和Ku80具有序列保守性,Ku70和Ku80在生物体内具有序列保守性,常被用来研究生物体内NHEJ途径,以及通过抑制其表达来提高基因敲除效率[20]。
圆红冬孢酵母(Rhodosporidiumtoruloides)的Ku70基因缺陷后,目标基因缺失频率到90%以上[21]。通过CRISPR技术敲低Ku70基因将猪胎儿成纤维细胞的同源重组的效率提高到1.85倍[22]。缺失Ku70可将斯达氏油脂酵母(LipomycesStarkeyi)基因打靶效率从10%提高到50%~100%[23]。敲除红色红曲菌M7的Ku70/80基因,同源重组效率提高2~3倍[24]。构建Ku70基因缺失菌株是提高红曲菌同源重组效率的有效途径。
本实验室前期对红曲菌的研究工作中基因敲除效率低于20%,因此本研究将通过敲除红曲菌中参与NHEJ途径的Ku70基因来提高红曲菌基因敲除效率,构建红曲菌的高效基因敲除体系,为红曲菌功能基因组学的研究工作提供优良菌株。
1 材料与方法
1.1 菌种和培养基
橙色红曲菌(Monascusaurantiacus)AS3.4384:购于中科院微生物菌种保藏中心,在4 ℃斜面MES培养基上保存。大肠杆菌DH5α:本实验室保存;农杆菌EHA105:中国科学院青岛生物能源与过程研究所赠送;质粒pCAMBIA1300:中国科学院青岛生物能源与过程研究所赠送;质粒pUC18-hph:本实验室构建。MES培养基:取500 g麦芽粉碎后加入2 L水,60 ℃糖化4 h,过滤收集滤液并煮沸,加入蛋清再次过滤,用水将滤液的波美度调至6,加入20 g·L-1琼脂粉,121 ℃灭菌15 min。MPPY液体培养基:40 g·L-1葡萄糖,2 g·L-1Yeast Extract,3 g·L-1NaNO3,0.5 g·L-1KCl,0.5 g·L-1MgSO4·7H2O,0.01 g·L-1FeSO4·7H2O,121 ℃灭菌15 min。G25N培养基:3 g·L-1NaNO3,1 g·L-1K2HPO4,0.5 g·L-1KCl,3 g·L-1NaNO3,0.5 g·L-1KCl,0.5 g·L-1MgSO4·7H2O,0.01 g·L-1FeSO4·7H2O,5 g·L-1Yeast Extract,30 g·L-1蔗糖,15g·L-1琼脂粉,121 ℃灭菌15 min。
1.2 Ku70蛋白的鉴定和同源性分析
以红色红曲菌Ku70蛋白序列(GenBank登录号:AGF90043.1)为同源序列,在橙色红曲菌AS3.4384的蛋白质组中进行同源搜索,将得到的Ku70蛋白序列提交至NCBI上,在Conserved Domains数据库中进行搜索,分析其保守的结构域并预测功能。
将红曲菌AS3.4384Ku70基因编码的氨基酸序列提交至NCBI数据库进行blastp分析,搜索与Ku70蛋白质序列同源性较高的生物序列,进行序列同源性比较。
1.3 pCAMBIA-△Ku70打靶载体的构建
使用NCBI分析红曲菌AS3.4384的核酸序列后,确定Ku70基因的位置及其上下游序列。根据重叠延伸PCR和无缝克隆的原理设计引物,并以AS3.4384基因组DNA为模板分别扩增Ku70 5’和Ku70 3’片段,以质粒pUC18-hph为模板扩增hph片段为抗性基因,通过重叠延伸PCR的方法将上述3个片段连接起来获得Ku70基因敲除盒。采用PCR的方法将载体pCAMBIA1300线性化。最后使用无缝克隆试剂盒将Ku70基因敲除盒与线性化载体连接,获得打靶载体pCAMBIA-△Ku70。打靶载体构建原理如图1所示,所需引物如表1所示。
1.4 农杆菌介导的红曲菌转化
红曲菌的转化借助农杆菌介导的T-DNA转化技术来实现[25]。出发菌株为AS3.4384,农杆菌为EHA105。转化过程中农杆菌EHA105的培养所用的培养基为含20 μg·mL-1利福平和30 μg·mL-1Kana的LB培养基,农杆菌EHA105介导的红曲菌转化所用的筛选平板为含150 μg·mL-1潮霉素和200 mM头孢霉素的MES平板。

图1 pCAMBIA-△Ku70打靶载体构建示意图Fig.1 Schematic diagram to construct pCAMBIA-△Ku70 targeting vector

表1 Ku70缺失菌株构建所需引物Tab.1 Primers of ku70-deficient strains were constructed
1.5 转化子的筛选及基因组DNA提取
挑取单菌落转化子到含150 μg·mL-1潮霉素的MES平板上,复筛转化子,28 ℃培养至菌落呈现红色,将其转接到5 mL MPPY液体培养基中,28 ℃,200 rpm培养至长出少量菌丝后,双层滤纸过滤,收集菌丝体,使用玻璃珠法提取基因组DNA[26]。
1.6 PCR验证转化子
以1.5中提取的转化子基因组DNA为模板,分别用287/288,289/507以及508/292三对引物,进行PCR验证,筛选出验证正确的Ku70基因缺失菌株。基因敲除原理如图2所示。

图2 红曲菌Ku70基因缺失菌株验证原理图Fig.2 Schematic diagram of validation of Monascus Ku70 gene deletion strain
1.7 单孢分离
将经PCR验证的候选转化子接种到含0.1% Triton X-100和150 μg·mL-1潮霉素的MES平板上,28 ℃,培养3 d;洗下闭囊壳,稀释后涂布在含0.1% Triton X-100和150 μg·mL-1潮霉素MES平板上,培养3 d;重复分离纯化一次;按1.5和1.6方法提取基因组DNA并进行PCR验证。
1.8 Ku70基因缺失菌株的生物学特性分析
分析Ku70基因缺失菌株(AS△Ku70)的生物学特性,研究AS△Ku70和起始菌株AS3.4384间的菌落形态、孢子产量以及红曲色素产量的差异变化。
1.8.1 菌落形态观察
在MES和G25N平板上分别接种2 μL 1×105个·mL-1的AS3.4384和AS△Ku70菌株的孢子悬浮液,28 ℃培养10 d,观察菌落大小及菌丝形态、颜色等表现型。
1.8.2 红曲菌孢子计数
挑取菌株AS3.4384和AS△Ku70菌丝到50 mL的MPPY液体培养基中,28 ℃,200 r·min-1,培养至菌丝开始产生色素。分别取1 mL菌液涂布到MES平板上,28 ℃培养16 d,加适量孢子洗液洗脱孢子,生理盐水洗涤两次,用血球计数板计数。洗下孢子后加热余下的培养基直至融化,过滤后纯水冲洗菌丝,收集菌丝体烘干后称重。
1.8.3 红曲色素的测定
接种105个AS3.4384和AS△Ku70孢子在含有50 g大米的大米培养基中,28 ℃静置培养,每天混匀两次,在第6、8、10、12、14、16和18 d取0.5 g样品,测其色素含量。色素的测定方法参考国标:《中华人民共和国国家标准 食品添加剂 红曲米GB4926-85》。
1.9 同源重组效率统计
基于同源重组原理和无缝克隆技术构建带有G418抗性基因的置换型Rab6和Rab17基因敲除打靶载体,然后借助1.4农杆菌转化法将打靶载体分别转入AS3.4384和AS△Ku70菌株,转化条件保持一致,根据1.5、1.6和1.7方法筛选并进行PCR扩增来验证转化子,统计转化子的同源重组效率。
2 结果
2.1 Ku70蛋白的鉴定及同源性分析
通过对Ku70蛋白结构域的分析(图3A),发现Ku70基因编码的蛋白是KU家族蛋白的一个亚基,KU家族的蛋白质参与非同源末端连接,用于修复双链DNA断裂。
将橙色红曲菌AS3.4384的Ku70基因编码的氨基酸序列提交至NCBI蛋白数据库进行比对,搜索与Ku70基因编码的氨基酸序列同源性较高的生物序列,进行多重序列比对,结果如图3B所示。Ku70基因编码的氨基酸序列与红色红曲菌(Monascusruber,GenBank:KC192955.1)、紫色红曲菌(Monascuspurpureus,GenBank:TQB71160.1)的同源性分别为96.43%、96.75%,说明Ku70蛋白在红曲菌属中高度保守。AS3.4384的Ku70蛋白与米曲霉(Aspergillusoryzae,GenBank:AB214649.1)、土曲霉(A.terreus,GenBank:GES65941.1)、黑曲霉(A.niger,GenBank:GAQ40800.1)的同源性分别为72.71%、73.12%、69.13%。
2.2 pCAMBIA-△Ku70打靶载体的构建
使用NCBI分析红曲菌AS3.4384的核酸序列后,确定Ku70基因的位置及其上下游序列。PCR扩增上游964 bp的序列为Ku70 5’(图4A泳道1)和下游961 bp的序列为Ku70 3’(图4A泳道3)。

图3 Ku70蛋白结构域分析(A)和Ku70蛋白多重序列比对图(B)Fig.3 Ku70 protein domain analysis (A) and multiple sequence alignment diagram of Ku70 protein
PCR扩增质粒pUC18-hph中3 060 bp的hph片段为抗性基因(图4A泳道2)。通过重叠延伸PCR的方法将三个片段连接起来,得到4 966 bp的Ku70基因敲除盒(图4A泳道4)。选择pCAMBIA1300为模板,采用PCR的方法获得6 800 bp的线性化载体(图4A泳道5)。
使用无缝克隆试剂盒将上述纯化后的目的片段和载体连接起来后,通过钙转感受态细胞的方法将连接产物转入DH5α感受态细胞中,挑取单菌落进行菌液PCR验证。首先使用引物473/474扩增Ku70基因敲除盒(4 966 bp,图4B泳道1~5),选出阳性克隆子并提取质粒。为进一步验证该阳性克隆子,以质粒为模板,使用引物473/474,327/473,329/328和474/330分别扩增Ku70基因敲除盒(4 966 bp,图4C泳道1)、Ku70 5’(947 bp,图4C泳道2)、hph(3 060 bp,图4C泳道3)和Ku70 3’片段(944 bp,图4C泳道4)。
2.3 红曲转化子的筛选和鉴定
借助农杆菌介导的T-DNA转化技术将pCAMBIA-△Ku70打靶载体转入出发菌株AS3.4384中,得到113个转化子,选取菌落颜色为红色的85个转化子进行复筛,培养3 d后,得到83个生长状态良好的转化子。选取其中36个转化子继续传代并进行单菌落分离,提取其基因组DNA,进行PCR验证。
以转化子基因组DNA为模板,使用3对引物287/288、289/507和508/292进行PCR验证。初次筛选确定30号转化子为AS△Ku70异核体菌株,扩增出Ku70 5’同源交换片段和Ku70 3’同源交换片段,但是也能扩增出Ku70缺失片段。对该菌株进行单孢分离后,再次进行PCR验证,转化子未能扩增出327 bp的Ku70缺失片段(图5A泳道1),但能扩增出3 642 bp的Ku70 5’同源交换片段(图5B泳道1)和3 172 bp的Ku70 3’同源交换片段(图5B泳道4),确定该转化子为AS△Ku70纯合子菌株。



(A) Ku70基因敲除载体构建。M:DL15000;泳道1:Ku70 5’;泳道2:hph片段;泳道3:Ku70 3’;泳道4:Ku70 5’-hph-Ku70 3’融合片段;泳道5:pCAMBIA1300线性化片段(B)打靶载体pCAMBIA-△Ku70转化子菌液PCR验证。M:DL15000;泳道1~5:转化子菌液;泳道6:阳性对照;泳道7:阴性对照;泳道8:空白对照(C)打靶载体pCAMBIA-△Ku70质粒PCR验证。M:DL15000;泳道1:Ku70 5’-hph-Ku70 3’融合片段;泳道2:Ku70 5’;泳道3:hph;泳道4:Ku70 3’
2.4 Ku70基因缺失菌株的生物学特性分析
2.4.1 菌落形态观察
将AS3.4384和AS△Ku70菌株分别接种到MES和G25N平板上,28 ℃培养10 d,观察菌落大小及菌丝形态、颜色等表现型差异。如图6A所示,AS△Ku70菌株在MES和G25N平板上的菌落大小与出发菌株AS3.4384基本一致,说明其生长速度与出发菌株基本没有差异,Ku70基因的缺失并不影响红曲菌的生长繁殖。AS△Ku70菌株的菌落形态与出发菌株相比无明显差距,说明Ku70基因的缺失并不影响红曲菌的菌体结构。
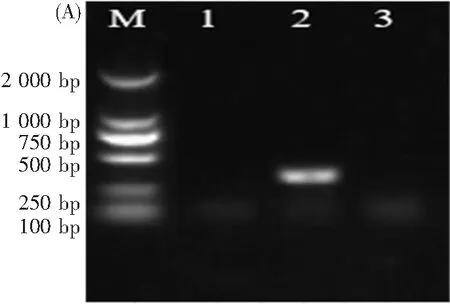
(A)红曲菌转化子Ku70缺失片段的PCR验证。M:DL2000;泳道1:转化子;泳道2:AS34384;泳道3:pCAMBIA-△Ku70质粒。(B)红曲菌转化子同源交换片段PCR验证。M:λ-HindⅢ;泳道1~3:转化子、AS34384、pCAMBIA-△Ku70质粒,扩增Ku705’端同源交换片段;泳道4~6:转化子、AS34384、pCAMBIA-△Ku70质粒,扩增Ku703’端同源交换片段。
2.4.2 红曲菌的孢子产量
将AS3.4384和AS△Ku70菌株在MES平板上培养16 d后,收集其孢子并使用血球计数板计数,同时测定菌丝体干重,将红曲菌产孢能力用每克菌丝体能够产生的孢子数量来衡量。如图6B所示,AS3.4384的产孢能力为2.26×107个·g-1,AS△Ku70的产孢能力为2.41×107个·g-1。表明AS△Ku70菌株与出发菌株AS3.4384相比其产孢能力和生长繁殖能力几乎无差距,Ku70基因的缺失并不影响红曲菌的产生孢子的能力和生长繁殖,可将AS△Ku70菌株作为出发菌株进一步敲除红曲菌其他基因。
2.4.3 红曲色素产量比较
将AS3.4384和AS△Ku70菌株接种到大米培养基中,28 ℃静置培养,每天混匀两次,在第6、8、10、12、14、16和18 d取0.5g样品,以红曲米提取液在420、470、505 nm下的吸光值来分别计算红曲菌株产黄色素、橙色素和红色素的产量,如图6C所示,培养18 d后,AS3.4384菌株的总色素产量为1304 U·g-1,AS△Ku70菌株的总色素产量为1 330 U·g-1,无明显差异。因此Ku70基因缺失不会影响红曲色素的生物合成。

(A)菌落形态比较
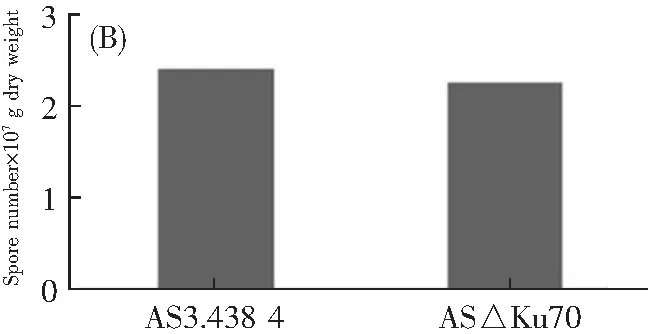
(B) 产孢能力

(C) 色素产量图6 Ku70基因缺失菌株的生物学特性分析Fig.6 Analysis of biological properties of Ku70 gene deletion strain
2.5 同源重组效率比较
以AS△Ku70菌株为起始菌株,AS3.4384菌株为对照菌株,使用相同的方法,分别敲除Rab6和Rab17基因,同源重组效率如表2所示。使用AS△Ku70菌株进行Rab6和Rab17基因敲除,转化子数量相对AS3.4384菌株减少约58%~65%,同源重组效率从0%~2.7%提高至15%~18.7%左右,提高了约6倍以上。

表2 AS3.4384和AS△Ku70的同源重组效率Tab.2 Homologous recombination efficiency of AS3.4384 and AS△Ku70
3 讨论
红曲菌作为传统的药食同源微生物,可产生多种有益的代谢产物和具有毒性的桔霉素[27]。构建基因敲除工程菌是研究红曲菌代谢途径和改造红曲菌株的有效方法[13-15]。红曲菌基因工程菌的构建过程包括打靶载体的构建、红曲菌的制备、转化和阳性转化子筛选及验证。该方法存在以下障碍:打靶载体复杂导致构建时间长、红曲菌含有刚性的细胞壁导致的转化效率低和同源重组发生概率低等。
打靶载体的常规构建方法是采用酶切酶连的克隆技术,该方法受限于酶切位点而不能随意在载体上插入目的基因,且实验周期长。本研究使用无缝克隆技术构建载体具有如下优势:可以不受载体酶切位点限制,将目的基因插入在所需要的位置;载体线性化可不依赖限制性内切酶,通过PCR的方式即可获得;连接载体与目的片段时不需要使用连接酶,直接用同源重组的方法20 min即可完成;阳性率高,减少了筛菌工作[28]。本研究构建打靶载体时需要借助无缝克隆不受载体酶切位点限制的优势,将Ku70 5’-hph-Ku70 3’融合片段与pCAMBIA1300载体上原有的hph基因进行置换。与普通克隆相比,本研究选择无缝克隆的方法构建pCAMBIA-△Ku70打靶载体缩短实验周期,提高工作效率。
常见的丝状真菌转化方法有聚乙二醇介导的原生质体转化、农杆菌介导的转化和电穿孔转化。聚乙二醇介导的转化和电穿孔转化都需要原生质体作为受体细胞,且对原生质体的质量要求高[29-30]。电穿孔转化还需要昂贵的仪器设备[31]。农杆菌介导的转化使用分生孢子即可,还具有高转化频率、高基因替换频率、T-DNA单拷贝整合等优点[32]。
本研究构建Ku70基因缺失菌株时曾尝试使用原生质体介导的原生质体转化,但由于得到的原生质体质量不佳、转化技术不到位等原因,重复若干次实验,原生质体再生率仅20%左右。转化后得到的转化子质量差,有的转化子产生红色色素却生长的气生菌丝较少,有的转化子铺满整个平板无法筛选出单菌落。改用农杆菌介导的转化后,仅做了两次实验便成功得到了阳性菌株。第一次实验失败的原因是筛选培养基中头孢霉素添加不足导致转膜后农杆菌泛滥生长抑制了红曲转化子的生长,第二次实验便成功得到了113个转化子,随机选取36个转化子进行分离鉴定成功筛选到阳性克隆子。故本研究选择农杆菌介导的转化也是提高红曲菌基因敲除效率的有效途径。
基因敲除效率与红曲菌体内的HR和NHEJ两种DNA双链断裂修复有关。由于HR仅在存在相同染色体作为模板的时候发生,而NHEJ不需要同源染色体,所以NHEJ是生物体内修复DNA双链断裂的主要途径,HR相对来说发生概率比较低[33]。有研究发现,Ku70蛋白是细胞核中的基于NHEJ途径的DNA修复因子,且NHEJ与HR在生物体内具有竞争性,可通过抑制NHEJ途径来提高HR途径的发生概率[34]。本研究构建了Ku70基因缺失菌株AS△Ku70,且分析AS△Ku70与AS3.4384的表现型及主要的代谢产物色素的产量的差异分析结果,证实Ku70基因的缺失对红曲菌的菌落形态、大小、产孢能力及红曲色素的代谢都几乎没有影响(图6)。分析AS△Ku70与AS3.4384的同源重组效率发现,Ku70基因缺失菌株的转化子数量减少约58%~65%,且同源重组效率提高约6倍以上。该结果可能与Ku70基因参与红曲菌的NHEJ途径相关,AS△Ku70中,发生NHEJ的效率降低,打靶载体随机插入的转化子数量减少,发生HR的转化子相对增加。后续可以通过优化转化条件提高AS△Ku70中的同源重组效率。
4 结论
本研究采用无缝克隆的技术手段,以橙色红曲菌AS3.4384为出发菌株,hph为抗性基因,构建了pCAMBIA-△Ku70打靶载体。借助农杆菌转化法将打靶载体转入出发菌株中,得到验证正确的突变体AS△Ku70。通过分析AS△Ku70菌株与AS34384菌株在菌落形态、产孢能力、代谢产物的产量及同源重组效率,得知Ku70基因并不参与红曲菌生长发育繁殖、初级代谢途径和次级代谢途径,且敲除Ku70基因同源重组效率提高6倍以上。AS△Ku70有望成为研究红曲菌代谢途径的优良菌株,为后续构建红曲菌的基因敲除工作提供便利。
