基于ESIPT的水杨醛吡啶衍生物发光调控机制的理论研究
2023-06-05赵建丽郭臻杨文静陈雪波
赵建丽,郭臻,杨文静*,陈雪波
(1.太原理工大学 材料科学与工程学院,山西,太原 030024;2.北京师范大学 理论计算光化学重点实验室,北京 100875)
0 引言
激发态分子内质子转移(ESIPT)反应是有机分子受光激发从基态跃迁到激发态,在激发态下分子内氢键被激活,其质子从酸性基团(OH 或NH2)转移到邻近的碱性基团(N=C 或C=O)发生质子转移互变异构反应的过程[1],ESIPT 反应具有大的斯托克斯位移和双荧光发射的特点,通常发生在具有分子内氢键的芳香族体系[2-4]。
水杨醛-4,6-(二甲基氨基)-吡啶(SDP)是一种典型的具有ESIPT 性质的有机小分子,其结构简单且易合成。国内外研究人员对SDP的合成和表征做了大量研究[5-6],Sun 团队通过缩合反应合成了SDP 及其衍生物,使用傅里叶红外光谱,氢谱,碳谱和质谱等表征手段研究了其光谱性质[7]。在此基础上,Teresa 团队利用电子吸收和荧光光谱对SDP 及其衍生物的光物理性质进行了评价,并采用含时密度泛函理论(TDDFT)研究了其前线分子轨道(MOs)和ESIPT 过程,进一步证实了该类分子具有苯酚向亚胺基吡啶环电荷转移的性质[8]。目前,对于这种水杨醛吡啶分子在激发态建立烯醇/酮式互变异构动态平衡导致双荧光发射的机制已有很多报道[9-13]。然而,针对取代基和溶剂对SDP 衍生物发光范围的调控效应还未展开深入研究,因此,迫切需要高精度的理论计算来解释这一问题。
如图1 所示,本文在SDP 的吡啶环亚胺基和苯环羟基对位分别引入氯原子,运用完全活化空间自洽场(CASSCF)[14-15]和完全活化空间二阶微扰(CASPT2)[16-17]方法,计算SDP 吡啶环亚胺基对位单取代氯原子(R-SDP-Cl)和苯环羟基对位单取代氯原子(L-SDP-Cl)的重要结构参数和红外线(IR)光谱,比较了二者的分子内氢键强度;模拟二者的静电势分布,分析了其质子迁移驱动力的大小;通过在气相、水、二甲亚砜、四氢呋喃和甲苯溶液中,获取到两种衍生物的ESIPT 能垒、吸收波长、发射能量、振子强度等重要光化学参数,来解释取代基和溶剂对ESIPT 过程的影响。此外,在溶剂效应下,结合激发态辐射速率公式,计算出R-SDPCl 和L-SDP-Cl 的荧光发射速率比,由此对其发光范围进行动力学评估。本文研究成果对SDP 这一类ESIPT 分子的设计及其发光范围评估提供了理性的参考设计方案。
1 计算方法
采用CASSCF[14-15]方法对R-SDP-Cl 和LSDP-Cl 进行稳态电子结构计算,并对这两个体系都选取了CAS(12e,10o)的活化空间。在选取活化轨道时,需要将涉及反应中心的轨道全部考虑进去。本文选取了N3 和Cl 的n 轨道、O1-H2 的σ/σ*轨道以及布局在整个共轭体系上离域程度不同的3 个占据π 轨道和3 个空轨道π*,构成了CAS(12e,10o)的活化空间(图2)。在CASPT2//CASSCF(12e,10o)/6-31G*理论水平下,计算了R-SDP-Cl 和L-SDP-Cl 的基态和激发态稳定点结构。
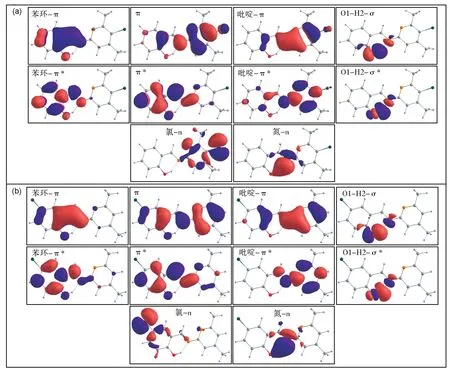
图2 R-SDP-Cl(a)和L-SDP-Cl(b)的活化轨道Fig.2 Active orbitals of R-SDP-Cl (a) and L-SDP-Cl (b)
基于高精度的CASSCF 方法,采用单根、两个根等权重(0.5,0.5)的态平均优化基态和激发态。对于(O1-H2···N3)质子迁移路径,采取固定键长优化的方法,即在给定的坐标下(O1-H2 的某一键长下),固定O1-H2 的键长,以此优化得到R-SDP-Cl 和L-SDP-Cl 的ESIPT 反应限制性势能曲线(CEPs)。CEPs 是以O1-H2 键长为横坐标,给定的键长下对应结构的能量为纵坐标绘制的最小能量曲线。为了考虑电子动态相关效应,这些稳定点在态平均CASSCF 方法优化的结构和轨道的基础上,采用CASPT2[16-17]方法进行单点能计算校正能量。为了考虑溶剂效应的影响,对两种衍生物在烯醇/酮式异构体相互转化过程中的关键点结构,即SCT(1ππ*)态上弛豫过程中出现的极小值和极大值,分别加入极化连续模型(PCM)和Hartee-Fock 半径的United Atom(UAHF)力场(水、二甲亚砜、四氢呋喃和甲苯)重新进行优化。基于优化的激发态稳定点结构,计算了两种衍生物的结构参数、IR 光谱图、静电势图以及势能曲线上的重要参数。为了探究溶剂效应对R-SDP-Cl 和L-SDP-Cl 发生ESIPT 反应、双荧光性质及发光范围的影响,对比了R-SDP-Cl和L-SDP-Cl 在气相与四种溶剂中ESIPT 反应路径和关键结构参数的变化,文中气相可以近似看作“理想气体状态”。此外,结合激发态辐射速率公式,计算出两种衍生物的荧光发射速率比,通过荧光发射速率比近似评估了两种衍生物的发光范围。本文计算结果均是基于CASPT2//CASSCF(12e,10o)/6-31G*理论水平上得到的。以上所有计算工作是通过Gaussian[18]程序结合Molcas[19]程序完成的。文中使用Multiwfn[20]程序绘制了R-SDP-Cl 和LSDP-Cl 静电势图。
对于分子的激发态辐射速率(kr)[21],使用下面公式对其进行简单估算。公式如下:
其中v为发射能量(s-1);e为元电荷(C);kr的单位是(s-1);ε0为真空介电常数;m为一个电子的质量;c是光速;fS1orT1→S0是S1→S0或T1→S0辐射跃迁的振子强度。
2 结果与讨论
2.1 基态和激发态分子内氢键
如图3 所示,R-SDP-Cl 的HOMO-LUMO轨道电荷分布呈镜像对称,因此可以判定它从基态向激发态的跃迁属于π-π*型。当电子从HOMO 轨道跃迁至LUMO 轨道后,R-SDP-Cl表现出明显的分子内电荷转移(ICT)特性,即电荷从羟基氧转移到亚胺基氮。同时,其偶极矩从基态(S0)的1.20 D 变到激发跃迁态(SCT(1ππ*))的4.00 D,这也进一步证明了该分子具有ICT 特性。通过上述分析,可以得出S0→SCT(1ππ*)激发具有电荷迁移性质。对L-SDP-Cl的MOs 和偶极矩的分析也得到了同样的结论。
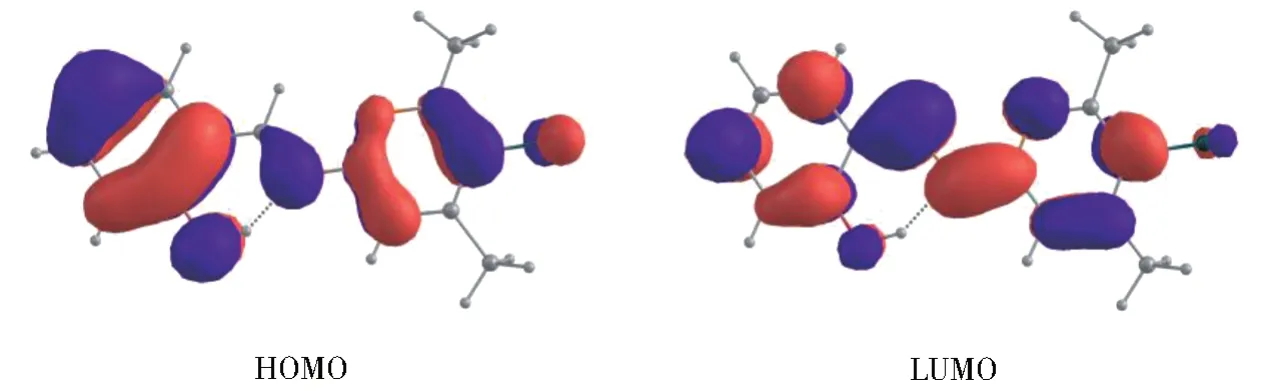
图3 R-SDP-Cl的前线分子轨道Fig.3 Frontier molecular orbitals of R-SDP-Cl
如表1 所示,优化得到了R-SDP-Cl 和LSDP-Cl 在S0态和SCT(1ππ*)态时烯醇式(Enol)和酮式(Keto)构型的分子内氢键键长(d)和键角(θ)。对于R-SDP-Cl 的Enol 构型,O1-H2 的键长从S0态的0.97 Å 增大到SCT(1ππ*)态的0.99 Å,H2···N3 的键长从S0态的1.87 Å 缩短到SCT(1ππ*)态的1.74 Å,O1···N3 的键长从S0态的2.72 Å 缩短到SCT(1ππ*)态的2.63 Å,此外,键角θ(O1-H2···N3)从S0态的144°增大到SCT(1ππ*)态的145 °。以上数据均表明,分子内氢键O1-H2···N3 在SCT(1ππ*)态得到加强,这有利于ESIPT 过程的发生[22]。对于LSDP-Cl 的Enol 构型,从S0态到SCT(1ππ*)态过程键长和键角的变化趋势与R-SDP-Cl 相似,这也表明L-SDP-Cl 的分子内氢键O1-H2···N3 在SCT(1ππ*)态得到加强,同样促进了ESIPT 过程的发生。

表1 R-SDP-Cl和L-SDP-Cl在Enol和Keto构型在S0和SCT(1ππ*)态的分子内H键的键长(Å)和键角(°)Table 1 Bond lengths (Å) and bond angles (°) of intramolecular hydrogen bonds of R-SDP-Cl and L-SDP-Cl in Enol and Keto configurations in S0 and SCT (1ππ*) states
同时,对于R-SDP-Cl 的Keto 构型,O1···H2 的长度从SCT(1ππ*)态的1.71 Å 缩短到S0态的1.64 Å,H2-N3 的长度从SCT(1ππ*)态的1.04 Å 增长到S0态的1.05 Å。此外,键角θ(O1···H2-N3)从SCT(1ππ*)态139°增加到S0态的141°。上述结果表明,O1···H2-N3 在S0态比SCT(1ππ*)态更稳定,这有利于基态的质子回迁。与R-SDP-Cl 的Keto 构型类似,L-SDP-Cl的Keto 构型的键长和键角也有相同的变化趋势,结果表明R-SDP-Cl 和L-SDP-Cl 的Keto 构型在S0态比在SCT(1ππ*)态更稳定。
另外,对电子激发状态下的氢键动力学特性也可以通过分子中相关化学键振动模式的光谱移动进行分析。R-SDP-Cl 和L-SDP-Cl 在S0态和SCT(1ππ*)态下O1-H2 键的红外伸缩振动频率图谱如图4 所示。R-SDP-Cl 和L-SDP-Cl的O1-H2 键伸缩振动频率从S0态的3553 cm-1和3564 cm-1红移至SCT(1ππ*)态的3196 cm-1和3065 cm-1,分别红移了357 cm-1和499 cm-1,这表明分子内氢键在SCT(1ππ*)态时得到了明显的增强,而且L-SDP-Cl 的增强程度更大,更有利于ESIPT 过程的发生。此外,取代基位置对O1-H2 键伸缩振动的影响主要反映在SCT(1ππ*)态,对S0态影响甚微。
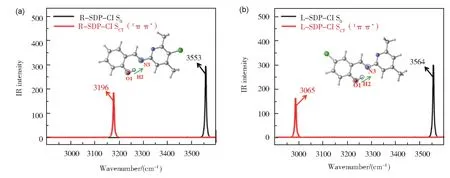
图4 R-SDP-Cl (a)和L-SDP-Cl (b)的烯醇式结构在S0态和SCT(1ππ*)态下O1-H2键的伸缩振动频率Fig.4 The calculated IR vibration spectra of enol form for R-SDP-Cl (a) and L-SDP-Cl (b) in S0 and SCT (1ππ*) states
2.2 分子表面静电势
R-SDP-Cl 和L-SDP-Cl 在Enol 的S0态和SCT(1ππ*)态的静电势分布如图5 所示,其中蓝色到红色代表分子表面静电势能逐渐升高,从颜色变化角度分析,从S0态到SCT(1ππ*)态颜色分布发生明显变化,分子内电荷发生很大程度的重排,这表明两种分子具有ICT 特性。从数值角度分析,位于R-SDP-Cl 羟基氧上的负电荷从S0态的33.56 kcal/mol 降低到SCT(1ππ*)态的20.89 kcal/mol,同时,亚胺基氮上的负电荷从S0态时的10.39 kcal/mol 升高到SCT(1ππ*)态的21.26 kcal/mol。羟基氧上的负电荷降低,可以削弱O1-H2 键的强度,亚胺基氮上的负电荷增加,可以吸引质子,进而促进ESIPT 过程的发生。对于L-SDP-Cl 的电荷分布分析也得到了类似的结论,这表明R-SDP-Cl 和LSDP-Cl 在SCT(1ππ*)态更容易发生ESIPT过程。

图5 R-SDP-Cl (a)和L-SDP-Cl (b)的Enol在S0态和SCT(1ππ*)态的静电势分布Fig.5 The ESP maps of R-SDP-Cl (a) and L-SDP-Cl (b) in S0 and SCT (1ππ*) states
此外,在SCT(1ππ*)态时,R-SDP-Cl 羟基氧上的静电势值为-20.89 kcal/mol,氨基氮上的静电势值为-21.26 kcal/mol,其静电势差值为0.37 kcal/mol,同理,L-SDP-Cl 的静电势差值为1.04 kcal/mol,这表明L-SDP-Cl 的质子迁移驱动力更大,更有利于ESIPT 过程的发生。
2.3 R-SDP-Cl和L-SDP-Cl的ESIPT反应路径
为了深入研究R-SDP-Cl 和L-SDP-Cl 的ESIPT 反应机制,由此绘制了两种衍生物在S0态和SCT(1ππ*)态沿质子转移方向能量变化的曲线图。如图6(a)所示,R-SDP-Cl 经过326 nm 的紫外光激发之后到达SCT(1ππ*)态的弗兰克-康登(FC)点,并很快通过一个快速衰变路径弛豫到极小点SCT-E,其能量比FC 点低了0.36 eV。这一初始弛豫在结构变化方面的最大特点是O1-H2 单键变弱。此时,SCT-E 辐射衰变到热基态,发射出约447 nm 蓝色荧光。在SCT(1ππ*)态上克服0.04 eV 的能垒后,沿着反应路线缓慢弛豫到达能量最低点SCT-K,SCT-K辐射衰变到热基态S0-K,发射出约565 nm 橘黄色荧光,之后S0-K 经过0.35 eV 能垒回到S0-E。L-SDP-Cl 经历类似的ESIPT 过程,正向反应能垒为0.02 eV,这表明L-SDP-Cl 更容易发生ESIPT 反应,这一结论与上述静电势和IR 光谱分析一致。

图6 R-SDP-Cl(a)和L-SDP-Cl(b)在气相中发生ESIPT反应最小能量路径Fig.6 The CEPs of ESIPT for R-SDP-Cl (a) and L-SDP-Cl (b) in the gas phase
基于CASPT2//CASSCF/6-31G* 理论水平,结合激发态辐射速率公式,计算得到RSDP-Cl 和L-SDP-Cl 在气相、水、二甲亚砜、四氢呋喃和甲苯溶剂环境中发生ESIPT 反应的吸收(发射)波长λ、振子强度f、辐射速率(kr1,kr2,s-1)以及速率比。从表2 可以看出,与RSDP-Cl 相比,L-SDP-Cl 的吸收(发射)波长发生明显的红移,使得L-SDP-Cl 产生近250 nm的Stokes 位移,这归因于氯原子的取代基效应。在基态时,对于L-SDP-Cl 而言,吸电子基氯原子处于苯环羟基的对位,使羟基氧原子负电荷减少(与静电势分布一致),因此L-SDP-Cl 的O1-H2 键被削弱,L-SDP-Cl 吸收较少的能量就可以到达激发态,从而导致吸收波长发生红移;当L-SDP-Cl 到达激发态后,因为L-SDP-Cl吸收的能量较少,因此其发射能量减少,使得L-SDP-Cl 的发射波长发生红移。与气相中相比,在极性较强的水和二甲亚砜中,R-SDP-Cl的吸收波长发生约5 nm 的蓝移,L-SDP-Cl 则发生约6 nm 的红移;在极性较弱的四氢呋喃和甲苯中,R-SDP-Cl 的吸收波长变化很小,而LSDP-Cl 发生红移,这表明取代基的位置与溶剂效应对吸收波长有一定的影响。
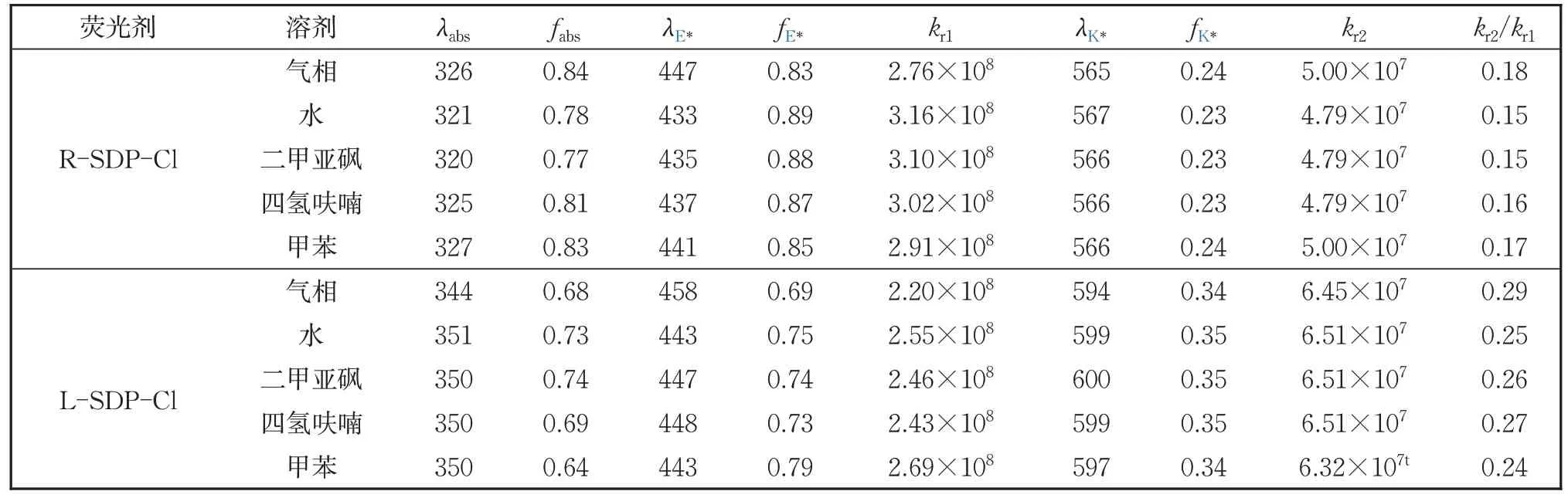
表2 计算得到R-SDP-Cl和L-SDP-Cl在气相、水、二甲亚砜、四氢呋喃和甲苯这五种氛围中,发生ESIPT反应的最大吸收(发射)波长λ、振子强度f、辐射速率(kr1、kr2,s-1)以及速率比Table 2 The maximum adsorption and emission wavelengths (nm), corresponding oscillator strengths (f) and the Radiation rate (kr1/kr2, s-1) as well as their ratio of ESIPT reaction for R-SDP-Cl and L-SDP-Cl in gas phase, H2O,dimethylsulfoxide, tetrahydrofuran and toluene solvents
2.4 R-SDP-Cl和L-SDP-Cl的发光范围
如表2 所示,当取代基氯原子的位置改变时,会影响R-SDP-Cl 和L-SDP-Cl 的荧光发射能和振子强度。将荧光发射能和振子强度代入激发态辐射速率公式,计算得到两种衍生物的蓝光发射速率kr1和黄光发射速率kr2。结果显示,R-SDP-Cl 的蓝光发射速率范围为2.76×108s-1~3.16×108s-1,黄光发射速率范围为4.79×108s-1~5.00×108s-1;L-SDP-Cl 的蓝光发射速率范围为2.20×108s-1~2.69×108s-1,黄光发射速率范围为6.32×108s-1~6.51×108s-1。这表明R-SDP-Cl 蓝光发射速率稍快于L-SDPCl,而黄光发射速率则相反。这是因为氯原子处于吡啶环亚胺基对位时,主要影响R-SDP-Cl和L-SDP-Cl 的keto 构型的电荷排布,由于氯原子和吡啶环的p-π 共轭效应,使得N3 上负电荷减少,从而影响H2 的迁移速率,降低了RSDP-Cl 发射黄光的振子强度,使得R-SDP-Cl的黄光发射速率变小;而氯原子处于苯环羟基对位时,主要影响R-SDP-Cl 和L-SDP-Cl 的enol 构型的电荷排布,由于氯原子的供电子效应,使得O1 上负电荷增多,从而束缚H2 的迁移,降低了L-SDP-Cl 发射蓝光的振子强度,使得L-SDP-Cl 的蓝光发射速率减小。由此,可以得出氯取代基对发光振子强度的影响较大,从而进一步影响蓝/黄光发射速率。
依据两种色光的发光二极管的配色原理[23],假设第一种单色发光二极管和第二种单色发光二极管的色坐标和亮度分别为(x1,y1,Y1)和(x2,y2,Y2),欲合成的目标颜色的色坐标和亮度(xm,ym,Ym),上述Y1,Y2是合成亮度为Ym时各单色光的发光亮度,因此,Ym=Y1+Y2。依据色度学原理和CIE1931 色彩图的定义,两种色光混合时,色光坐标到合成点距离比等于色光光量的反比(结合刺激纯度和亮度纯度的定义),得到如下公式:
联立(2)、(3)式可得(4)和(5),
两种色光的亮度比可用式(6)表示:
其中x1,y1,x2,y2,xm,ym的值为给定值,K2可根据式(4)或(5)推导得出,因此两种色光的配比可根据目标色估算得到。但是CIE 色坐标几乎不受发光强度的影响,式(6)中没有考虑两种色光发光强度的影响。假设两种色光的原始发光强度分别为Y10,Y20,并假设Y10最小,那么两种色光的辐射速率比被修正为式(7):
依据式(4)、(5)和(7),结合表2 中的kr2/kr1以及蓝色光的CIE(0.15,0.07)和黄色光的CIE(0.40,0.52)(CIE 值从CIE1931 色彩图中获得),可以计算得到蓝色光和黄色光混合之后的混合光的CIE 值,以此来近似评估混合光的发光质量和范围。如图7 所示,计算得到了两种衍生物混合光的CIE(xm,ym),由于数据比较密集,因此,只绘制了两种衍生物在气相中的CIE 图。计算结果发现,R-SDP-Cl 的混合光的发光范围为CIE(0.138,0.083)~CIE(0.139,0.086),L-SDP-Cl 的混合光的发光范围为CIE(0.142,0.090)~CIE(0.145,0.095)。对照CIE1931 色彩图可知,两种衍生物最后发射出单一蓝光,且L-SDP-Cl 蓝光范围的拓展程度更大。通过以上分析,发现L-SDP-Cl 的蓝光范围要比R-SDP-Cl 更宽一些,且周围溶剂环境对其发光范围的影响甚微,因此,对发光范围起主要影响作用的是取代基。这些调控理论为OLED 材料分子设计和发光范围调控提供合理化方案。

图7 R-SDP-Cl和L-SDP-Cl在气相中的CIE图及水、二甲亚砜、四氢呋喃和甲苯溶剂中的CIE值Fig.7 CIE diagrams of R-SDP-Cl and L-SDP-Cl in gas phase and CIE values in water, dimethyl sulfoxide,tetrahydrofuran and toluene solvents
3 结论
本文在高精度CASPT2 和CASSCF 方法的基础上结合激发态辐射速率公式,研究了RSDP-Cl 和L-SDP-Cl 在气相、水、二甲亚砜、四氢呋喃和甲苯溶液中的ESIPT 反应路径及发光调控机制。结果表明,当R-SDP-Cl 和LSDP-Cl 受光激发后,分子内电荷发生明显重排,即电子由羟基氧转移到亚胺基氮,为ESIPT反应的发生提供驱动力;分子在SCT(1ππ*)态建立烯醇/酮式互变异构动态平衡过程中以一定速率发射蓝光和黄光,最后混合得到单一蓝光;当吡啶环亚胺基和苯环羟基对位分别引入氯原子后,取代基效应会影响ESIPT 反应的能垒大小,但能垒差仅为0.02 eV,因此,其ESIPT反应接近于无能垒过程。根据两种色光的发光二极管的配色原理,将荧光辐射速率比类比于亮度比,近似评估两种衍生物的发光范围,计算预测出L-SDP-Cl 对发光范围的拓展效果更好。研究发现取代基可以调控发光范围,且周围溶剂环境也可以在一定程度上对其进行微调。本工作不仅为SDP 衍生物分子的ESIPT反应机制提供了清晰的理论模型,而且还为OLED 材料分子发光范围的调控提供了具有实用价值的方案。
