锈毛钝果寄生叶绿体基因组的序列特征和系统发育分析
2023-05-20王军峰汤紫依何海叶张慧娟
蒋 明,王军峰,吴 丹,朱 晏,汤紫依,何海叶,张慧娟*
锈毛钝果寄生叶绿体基因组的序列特征和系统发育分析
蒋 明1,王军峰2,吴 丹3,朱 晏1,汤紫依1,何海叶1,张慧娟1*
1. 台州学院生命科学学院,浙江 椒江 318000 2.华东药用植物园科研管理中心,浙江 丽水 323000 3. 台州恩泽医疗中心(集团)路桥医院,浙江 路桥 318050
以药用植物锈毛钝果寄生为材料,在高通量测序、组装的基础上,明确叶绿体因组的结构、序列特征和系统发育关系。利用SDS法提取基因组DNA,采用Illumina HiSeq X Ten进行高通量测序,用NovoPlasty组装叶绿体基因组,借助PhyML生成系统发育树。锈毛钝果寄生的叶绿体基因组全长为122 208 bp,GC值为37.3%,大单拷贝区(large single copy region,LSC)、小单拷贝区(small single copy region,SSC)和反向重复区(inverted repeat region,IR)的长度分别为70 522、6084和22 801 bp;锈毛钝果寄生的叶绿体基因组共有基因108个,其中的编码蛋白基因、tRNA与rRNA分别为66、29和8个,另有5个假基因;基因、所有的基因及6个tRNA发生丢失。序列比对结果表明,锈毛钝果寄生与桑寄生(Lecomte) Danser叶绿体基因组之间的相似性最高,达96.7%。系统发育分析结果表明,7种植物在发育树上分成5组,其中,锈毛钝果寄生与桑寄生和广寄生聚于一组。锈毛钝果寄生叶绿体基因组的组装、序列分析和系统发育分析,为后续开展遗传结构和遗传多样性研究奠定了基础,并为桑寄生科植物的进化和系统发育研究提供了依据。
锈毛钝果寄生;桑寄生;广寄生;叶绿体基因组;序列特征;系统发育分析
寄生植物(Parasitic plants)是植物界的特殊类群,全世界约有4500种,它们营全寄生或半寄生生活,通过吸器从寄主中获取生长发育所需要的物质[1]。全寄生植物(Holoparasitic plants)由于不能进行光合作用,所有的养分都来自寄主;半寄生植物(Hemiparasitic plants)不完全依靠寄主获得营养,它们的叶片具有叶绿体,能通过光合作用制造养分[2-3]。大花草科(Rafflesiaceae)、檀香科(Santalaceae)、列当科(Orobanchaceae)、锁阳科(Cynomoriaceae)、蛇菰科(Balanophoraceae)和桑寄生科(Loranthaceae)为寄生植物的几个主要科,其中的大部分植物为半寄生[4-7]。桑寄生科植物为半寄生性灌木、亚灌木或草本,寄生于木本植物的根、茎或枝条上,全世界约65属,1300余种,我国有64种,10变种[8]。
锈毛钝果寄生(Merr.) H. S. Kiu为桑寄生科钝果寄生属灌木,分布于广西、云南、湖南、安徽和浙江等省,主要寄生于油茶Abel.、樟(L.) Presl和壳斗科Fagaceae植物[8]。锈毛钝果寄生的叶片、嫩枝、花蕾和花的表面密被星状毛,这一特征可与同属其他物种相区分(图1)。锈毛钝果寄生具有一定的药用价值,全株入药,有祛风除湿功效[8]。锈毛钝果寄生在江苏省也有发现,该植物随灰毛含笑var.Law et Y. F. Wu的引种带入[9]。有关锈毛钝果寄生的研究很少,仅见于化学成分和新记录种等方面的研究[10-11]。叶绿体基因组是独立于核基因组的小型环状DNA分子,它具有结构保守、基因数量和排列稳定等特点,在保护生物学、遗传多样性、基因水平转移和物种鉴定等方面取得了一些进展[12-16]。目前,有关锈毛钝果寄生叶绿体基因组方面的研究未见报道,本研究在测序和组装的基础上,对锈毛钝果寄生叶绿体基因组进行序列分析和系统发育分析,为后续开展该物种的遗传结构和遗传多样性研究奠定基础;同时,比较了锈毛钝果寄生、桑寄生(Lecomte) Danser和广寄生(DC.) Danser的叶绿体基因组序列,并为桑寄生科植物的进化和系统发育提供依据。

A-枝条和叶片 B-花
1 材料与仪器
1.1 材料
锈毛钝果寄生的叶片采自浙江丽水碧湖,寄主植物为枫杨C. DC.,采集健康、幼嫩的叶片,置于取样袋。叶片带回实验室后,用无菌水冲洗3~5次,晾干后置于−80 ℃冰箱备用。
1.2 仪器
ThinkPad P52移动工作站(联想有限公司);超净工作台(苏州安泰空气技术有限公司);Covaris超声波DNA破碎仪(Chromatin Shearing公司,美国);伯乐C1000型PCR仪(Bio-Rad公司,美国);Illumina HiSeq X Ten测序仪;DYY-12型电泳仪及电泳槽(北京市六一仪器厂);伯乐Gel Doc XR+凝胶成像系统(Bio-Rad公司,美国)。
2 方法
2.1 DNA的提取和测序
叶片用液氮快速冷冻后研磨成粉末,采用SDS法提取基因组DNA。利用Covaris超声波破碎仪制备长度为350 bp的DNA片段,经末端修复、加尾、加接头、纯化和PCR扩增等步骤完成测序文库的构建。测序在Illumina HiSeq X Ten上进行,策略为双末端(Paired-End,PE)PE150测序。
2.2 叶绿体基因组的组装和边界验证
高通量测序得到6.75 G原始数据,去除接头和低质量的数据区,获得clean reads 27 130 560条。利用NOVOPlasty拼接叶绿体基因组,采用默认参数[17]。用于边界验证的PCR引物分别为:P1:5’-CCGTATGCTTTGGAAGAAGCT-3’、P2:5’-GGCCTGTAGTAGGTATCTGGTTCAC-3’、P3:5’-CTCCCAATTTGTGACCTACCATACG-3’、P4:5’-GACGGAACGGGAAGACCTAGG-3’、P5:5’-GGCAGAATACCATCGCCCATTC-3’、P6:5’-CCTGTTAGACAGCAAAATCCCGC-3’、P7:5’-GCCACCTCTTCGGTATTTGTCTG-3’、P8:5’-AGGCAGAATACCATCGCCCA-3’。PCR反应采用20 μL体系,分别加入ddH2O 15.5 μL、10×缓冲液2.0 μL、10 mmol/L的dNTPs 0.6 μL、20 μmol/L的上游引物/下游引物各0.4 μL、50 ng/μL的模板0.6 μL和2 U/μL酶0.5 μL。片段克隆在伯乐C1000型PCR仪上进行,程序为:94 ℃预变性5 min;94 ℃变性30 s,54~56 ℃退火45 s,72 ℃退火1 min,共32个循环;72 ℃最后延伸10 min。PCR产物经电泳检测、割胶回收、连接和转化,取阳性克隆测序。
2.3 基因组的注释和比较
锈毛钝果寄生叶绿体基因组的注释采用DOGMA(Dual Organellar GenoMe Annotator)程序(http://dogma.ccbb.utexas.edu/),并经手工调整[18];tRNA的注释采用tRNAscan-SE (http://lowelab. ucsc.edu/tRNAscan-SE/)和ARAGORN工具[19-20];利用Organellar Genome DRAW生成圈图[21]。
2.4 系统发育分析
从NCBI下载6种植物的叶绿体基因组序列,它们分别来自桑寄生科钝果寄生属的桑寄生(登录号:KY996493)、广寄生(KY996492)、梨果寄生属L.的红花寄生L.(NC_040862)、鞘花属L.的鞘花(Lour.) Van Tiegh.(NC_039376)、离瓣寄生属Lour.的离瓣寄生Lour.(NC_039375)和菊科(Compositae)的水飞蓟(L.) Gaertn.(KT267161)等的叶绿体基因组序列,其中的水飞蓟序列用作外类群。Mauve插件用于钝果寄生属植物叶绿体基因组的比对,参数采用默认值。jModelTest工具用于筛选核苷酸替代模型,利用PhyML软件生成最大似然树[22]。
3 结果与分析
3.1 锈毛钝果寄生叶绿体基因组的结构
利用NOVOPlasty完成了clean reads的拼接,并对边界序列进行PCR克隆和测序验证。结果表明,锈毛钝果寄生叶绿体基因组的全长为122 208 bp,GC值为37.3%,该基因组具有1个典型的四分体结构,即整个环状基因组可分成大单拷贝区(large single copy region,LSC)、小单拷贝区(small single copy region,SSC)及2个反向重复区(inverted repeat region,IR)(图2)。LSC、SSC和IR的长度分别为70 522 bp、6 084 bp和22 801 bp,它们的GC值分别为34.7%、26.4%和42.9%。

图2 锈毛钝果寄生叶绿体基因组
3.2 基因组成
锈毛钝果寄生叶绿体基因组共有108个基因,包括29个tRNA、8个rRNA、66个编码蛋白基因和5个假基因(表1)。tRNA中,、、、和各有2份拷贝,其中的具1个内含子。rRNA共有4种,每种2份拷贝,它们分布于IR区域。编码蛋白基因中,核糖体蛋白大亚基、核糖体蛋白小亚基和及未知功能蛋白各有2份拷贝;、、、和均有1个内含子,而和各有2个内含子。、和为假基因,其中的和各有2份拷贝。锈毛钝果寄生叶绿体基因组中,所有基因,即全部缺失,tRNA基因、、、、和也发生缺失。此外,在锈毛钝果寄生叶绿体基因组中未能检测到翻译起始因子基因。

表1 锈毛钝果寄生叶绿体基因组的基因
×2-拷贝数为2 Ψ-假基因 *-1个内含子 **-2个内含子
× 2-two copies Ψ-pseudogene *-one intron **-two introns
3.3 与同属植物的全基因组比较
广寄生、桑寄生和锈毛钝果寄生为同属植物,它们均营半寄生生活。广寄生与桑寄生的叶绿体基因组全长分别为121 363 bp和122 562 bp,而锈毛钝果寄生则介于两者之间,3种植物叶绿体基因组的GC值均为37.3%。3种植物叶绿体基因组的基因结构和基因组成相似,它们的基因数均为108,其中66个为蛋白编码基因,37个为RNA。锈毛钝果寄生与桑寄生叶绿体基因组之间的相似性较高,达96.7%,与广寄生的相似性略低,为92.7%(图3)。序列之间的差异主要表现在基因间隔区,如-、和间隔区存在大量的插入/缺失现象。编码蛋白基因之间也存在一定的差异,如常用的叶绿体DNA标记中,3种植物的基因之间存在68个变异位点,导致40个氨基酸残基产生差异(图4);和的DNA序列各有40个变异位点;而基因相对保守,仅4个变异位点。

图3 3种钝果寄生属植物叶绿体基因组的比较
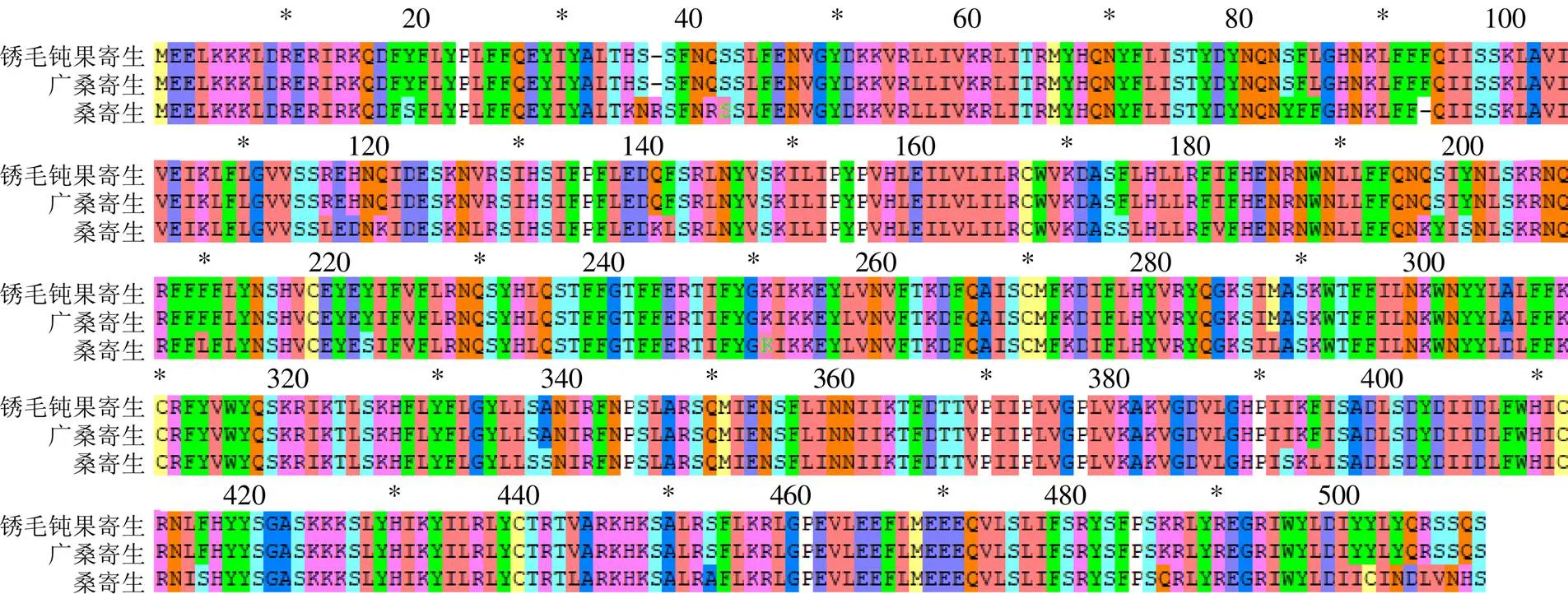
图4 3种钝果寄生属植物matK蛋白序列的比较
3.4 系统发育分析
利用jModelTest工具获得分子进化模型,预测结果表明,GTR+G+I为最佳替代模型,赤池信息标准(Akaike information criterion,AIC)与贝叶斯信息标准(Bayesian information criterion,BIC)分别为512 130.246 98和512 316.620 09。7种植物的叶绿体基因组在系统发育树上可分为5组,同为钝果寄生属的广寄生、桑寄生和锈毛钝果寄生聚为1组(II),梨果寄生属的红花寄生(I)、鞘花属的鞘花(III)、离瓣寄生属的离瓣寄生(IV)及外类群水飞蓟(V)各占1个分支(图5)。锈毛钝果寄生和桑寄生的序列差异最小,遗传距离最近,两者处于同一分支,支持率达100%;它们再与广寄生聚为1组,支持率为100%。
4 讨论
叶绿体是植物进行光合作用的场所,也是氮、硫同化,氨基酸、叶绿素、类胡萝卜素及脂肪酸合成的重要部位[23]。叶绿体基因组呈环状,通常可分成4个部分,即LSC、SSC和2个反向重复区IR。但也有例外,在松科(Pinaceae)和丝藻属Friedl植物中,IR的1个拷贝发生缺失[24-25]。在陆生植物中,叶绿体基因组序列的长度通常为120~160 kb,由70~88个蛋白质编码基因及33~35个RNA基因组成[26]。绿色植物的叶绿体基因组较大、基因数量较多,枸杞Miller叶绿体基因组的全长为155 756 bp,共有130个基因,其中编码蛋白基因的数量为85个,tRNA与rRNA基因分别为30个和8个[27];金荞麦(D. Don) Hara的叶绿体基因组全长为159 919 bp,共有131个基因,包括80个蛋白质编码基因、28个tRNA基因和8个rRNA基因[28];水飞蓟(L.) Gaertn.叶绿体基因组的大小为153 202 bp,有137个基因,其中蛋白质编码基因89个,tRNA基因40个,rRNA基因8个[29]。而非绿色植物中的叶绿体基因组显著变小,基因数量则明显减少,本研究中,锈毛钝果寄生叶绿体基因组全长仅为122 208 bp,基因数量仅108个,包括66个蛋白质编码基因,29个tRNA和8个rRNA。与同属的桑寄生和广寄生相比,它们的基因组成和基因结构十分相似,但序列上存在较多的差异,这些差异在基因和基因间隔区均有发现,它们可用于3种植物的分子鉴定。
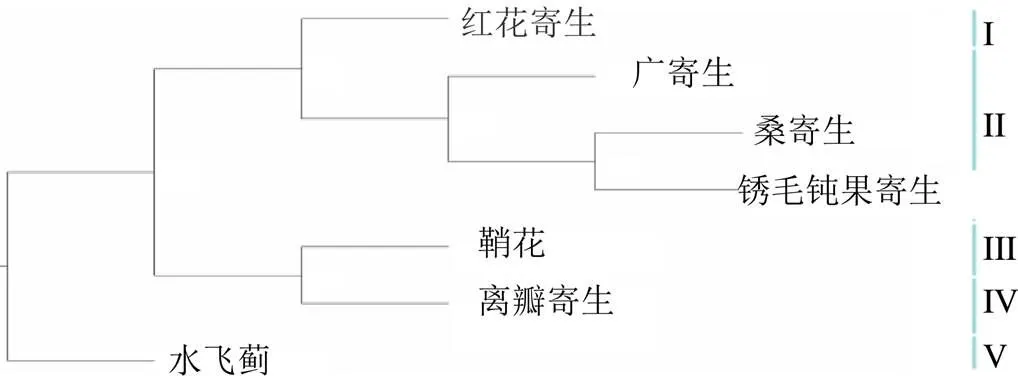
图5 基于叶绿体基因组序列构建的系统发育树
与陆生非寄生植物相比,寄生植物基因丢失现象十分频繁。山毛榉寄生(L.) Bartram为全寄生植物,通常寄生于大叶山毛榉Ehrh.的根部,该植物的叶绿体基因组全长为70 028 bp,仅含42个基因,一半为蛋白质编码基因[30]。肉苁蓉Y. C. Ma的叶绿体基因组大小为102 657 bp,含60个基因,其中编码蛋白基因27个[31]。Molina等[32]以全寄生植物大王花Blanco的DNA为材料,检测到17个蛋白质编码基因、2个rRNA和3个tRNA的基因片段,但无法拼接出完整的叶绿体基因组,推测该植物的叶绿体基因组已丢失。半寄生植物叶绿体中也有类似的基因组变小、基因丢失等现象。桑寄生与广寄生的叶绿体基因组大小为121 363 bp和122 562 bp,它们各有106个基因,其中的蛋白编码基因、tRNA及rRNA基因的数量分别为66、28和8,另有4个假基因[33]。与桑寄生和广寄生相比,钝果锈毛寄生的叶绿体基因多2个,但编码蛋白基因数量一致,也为66个。大苞寄生(Merr.) Danser叶绿体基因组较钝果寄生属植物稍大,为123 581 bp,共有119基因,其中77个为编码蛋白基因[34]。
在叶绿体基因组进化过程中,假基因化是一种较为常见的现象,序列变异、IR区域的扩张(expansion)和收缩(contraction)等是造成叶绿体基因组假基因化的原因[35]。金匙木属L.植物中,IR变界部位的和为假基因[35];芝麻L.叶绿体基因组中也有类似的现象,即IR变界处的和分别假基因化[36]。肉苁蓉叶绿体基因组的假基因数量较多,共有24个,如、、、、和等[31]。而半寄生植物中,假基因化现象相对较少,桑寄生和广寄生中,、和为假基因,其中的有2份拷贝[33]。与桑寄生和广寄生类似,锈毛钝果寄生的、和也发生假基因化,但锈毛钝果寄生中,有2份拷贝。寄生植物叶绿体基因组基因丢失现象也十分普遍,山毛榉寄生叶绿体中所有的RNA聚合酶基因及大部分tRNA和rRNA丢失[30]。肉苁蓉叶绿体基因组的基因全部缺失,但保留了所有的tRNA基因[31]。与肉苁蓉略有不同,锈毛钝果寄生叶绿体基因组所有基因也发生丢失,并且它的tRNA成员不完整,、、、、和完全丢失。基因的编码蛋白NDH参与光系统I的电子传递,在光合反应过程中起着重要作用,而的丢失,将影响锈毛钝果寄生的光合作用,基因丢失与寄生习性密切相关。另外,在锈毛钝果寄生叶绿体基因组中未能检测到翻译起始因子基因,类似现象出现于大叶胡颓子Thunb.、丝瓣剪秋罗(Regel) Maxim.、头序蝇子草Kom.、细果野菱Korsh.和福氏紫薇Koehne等植物[37-39]。水平转移在很多被子植物中均有发现,锈毛钝果寄生中是否存在这一现象,尚需进一步验证[40]。
本研究以半寄生植物锈毛钝果寄生为材料,在高通量测序的基础上,对叶绿体基因组进行了拼接和边界验证,并开展了序列特征与系统发育分析,为后续开展该物种的遗传结构和遗传多样性研究奠定了基础。
利益冲突 所有作者均声明不存在利益冲突
[1] Těšitel J. Functional biology of parasitic plants: A review [J]., 2016, 149(1): 78-89.
[2] DePamphilis C W, Young N D, Wolfe A D. Evolution of plastid gene rps2 in a lineage of hemiparasitic and holoparasitic plants: Many losses of photosynthesis and complex patterns of rate variation [J]., 1997, 94(14): 7367-7372.
[3] McKibben M, Henning J A. Hemiparasitic plants increase alpine plant richness and evenness but reduce arbuscular mycorrhizal fungal colonization in dominant plant species [J]., 2018, 6: e5682.
[4] Press M C, Phoenix G K. Impacts of parasitic plants on natural communities [J]., 2005, 166(3): 737-751.
[5] Cusimano N, Renner S S. Sequential horizontal gene transfers from different hosts in a widespread Eurasian parasitic plant,[J]., 2019, 106(5): 679-689.
[6] Sanchez-Puerta M V, Edera A, Gandini C L,. Genome-scale transfer of mitochondrial DNA from legume hosts to the holoparasite(Balanophoraceae) [J]., 2019, 132: 243-250.
[7] Ornelas J F, García J M, Ortiz-Rodriguez A E,. Tracking host trees: The phylogeography of endemic(Loranthaceae) mistletoe in the Sonoran Desert [J]., 2019, 110(2): 229-246.
[8] 中国科学院中国植物志编辑委员会. 中国植物志(第七十二卷)[M]. 北京: 科学出版社, 1988: 119-136.
[9] 叶康. 江苏种子植物分布新记录科: 桑寄生科 [J]. 种子, 2015, 34(3): 58-59.
[10] 李良琼, 李美蓉, 朱爱江. 锈毛寄生化学成分的研究 [J]. 中国中药杂志, 1996, 21(1): 34-35.
[11] 丘华兴, 陈炳辉, 曾飞燕. 值得注意的中国南部植物 [J]. 广西植物, 2006, 26(1): 1-4.
[12] Wambugu P W, Brozynska M, Furtado A,. Relationships of wild and domesticated rices (AA genome species) based upon whole chloroplast genome sequences [J]., 2015, 5: 13957.
[13] Brozynska M, Furtado A, Henry R J. Genomics of crop wild relatives: Expanding the gene pool for crop improvement [J]., 2016, 14(4): 1070-1085.
[14] Daniell H, Lin C S, Yu M,. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering [J]., 2016, 17(1): 134.
[15] Bi Y, Zhang M F, Xue J,. Chloroplast genomic resources for phylogeny and DNA barcoding: A case study on[J]., 2018, 8(1): 1184.
[16] 赵月梅, 杨振艳, 赵永平, 等. 木犀科植物叶绿体基因组结构特征和系统发育关系 [J]. 植物学报, 2019, 54(4): 441-454.
[17] Dierckxsens N, Mardulyn P, Smits G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data [J]., 2017, 45(4): e18.
[18] Wyman S K, Jansen R K, Boore J L. Automatic annotation of organellar genomes with DOGMA [J]., 2004, 20(17): 3252-3255.
[19] Lowe T M, Eddy S R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence [J]., 1997, 25(5): 955-964.
[20] Laslett D, Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences [J]., 2004, 32(1): 11-16.
[21] Lohse M, Drechsel O, Kahlau S,. OrganellarGenomeDRAW: A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets [J]., 2013, 41(Web Server issue): W575-W581.
[22] Guindon S, Dufayard J F, Lefort V,. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0 [J]., 2010, 59(3): 307-321.
[23] Jensen P E, Leister D. Chloroplast evolution, structure and functions [J]., 2014, 6: 40.
[24] Wu C S, Wang Y N, Hsu C Y,. Loss of different inverted repeat copies from the chloroplast genomes of Pinaceae and cupressophytes and influence of heterotachy on the evaluation of gymnosperm phylogeny [J]., 2011, 3: 1284-1295.
[25] Turmel M, Otis C, Lemieux C. Mitochondrion- to-chloroplast DNA transfers and intragenomic proliferation of chloroplast group II introns ingreen algae (Ulotrichales, Ulvophyceae) [J]., 2016, 8(9): 2789-2805.
[26] Wicke S, Schneeweiss G M, DePamphilis C W,. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function [J]., 2011, 76(3/4/5): 273-297.
[27] Yang Z R, Huang Y Y, An W L,. Sequencing and structural analysis of the complete chloroplast genome of the medicinal plantmill [J].(), 2019, 8(4): 87.
[28] Wang X M, Zhou T, Bai G Q,. Complete chloroplast genome sequence of: Genome features, comparative analysis and phylogenetic relationships [J]., 2018, 8(1): 12379.
[29] Zhang Y, Li B, Chen H,. Characterization of the complete chloroplast genome of(Sapindales: Aceraceae), a rare and vulnerable tree species endemic to China [J]., 2016, 8(4): 383-385.
[30] Wolfe K H, Morden C W, Palmer J D. Function and evolution of a minimal plastid genome from a nonphotosynthetic parasitic plant [J]., 1992, 89(22): 10648-10652.
[31] Li X, Zhang T C, Qiao Q,. Complete chloroplast genome sequence of holoparasite(Orobanchaceae) reveals gene loss and horizontal gene transfer from its host(Chenopodiaceae) [J]., 2013, 8(3): e58747.
[32] Molina J, Hazzouri K M, Nickrent D,. Possible loss of the chloroplast genome in the parasitic flowering plant(Rafflesiaceae) [J]., 2014, 31(4): 793-803.
[33] Li Y, Zhou J G, Chen X L,. Gene losses and partial deletion of small single-copy regions of the chloroplast genomes of two hemiparasiticspecies [J]., 2017, 7(1): 12834.
[34] Yu R X, Zhou S Y, Zhou Q J,. The complete chloroplast genome of a hemiparasitic plant(Loranthaceae) [J]., 2019, 4(1): 207-208.
[35] Menezes A P A, Resende-Moreira L C, Buzatti R S O,. Chloroplast genomes ofspecies (Malpighiaceae): Comparative analysis and screening of high divergence sequences [J]., 2018, 8(1): 2210.
[36] Zhang H Y, Li C, Miao H M,. Insights from the complete chloroplast genome into the evolution ofL [J]., 2013, 8(11): e80508.
[37] Choi K S, Son O G, Park S. The chloroplast genome ofand trnH duplication event in Elaeagnaceae [J]., 2015, 10(9): e0138727.
[38] Kang J S, Lee B Y, Kwak M. The complete chloroplast genome sequences ofandand comparative analyses with other Caryophyllaceae genomes [J]., 2017, 12(2): e0172924.
[39] Xue Z Q. The complete chloroplast DNA sequence ofKorsh. (Trapaceae), and comparative analysis with other Myrtales species [J]., 2017, 143: 54-62.
[40] Millen R S, Olmstead R G, Adams K L,. Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus [J]., 2001, 13(3): 645-658.
Chloroplast genome sequence characterization and phylogenetic analysis of
JIANG Ming1, WANG Jun-feng2, WU Dan3, ZHU Yan1, TANG Zi-yi1, HE Hai-ye1, ZHANG Hui-juan1
1. College of Life Sciences, Taizhou University, Jiaojiang 318000, China 2. Scientific Research Management Center, East China Medicinal Botanical Garden, Lishui 323000, China 3. Luqiao Hospital, Taizhou Enze Medical Center (Group), Luqiao 318050, China
In the present study, based on high-throughput sequencing and genome assembly methods, chloroplast genome structure, sequence characters, and phylogenetic relationships ofwere confirmed.The SDS-based DNA extraction method was applied to prepare genomic DNA, and the Illumina HiSeq X Ten System was used for high-throughput sequencing. NovoPlasty was chosen to assemble chloroplast genome. A phylogenetic tree was generated using PhyML.The complete chloroplast genome ofwas 122 208 bp in length with a GC content of 37.3%, and the sizes of LSC, SSC, and IR were 70 522 bp, 6 084 bp, and 22 801 bp, respectively. The chloroplast genome harbored 108 genes, and the numbers of protein-coding genes, tRNAs, and rRNAs were 66, 29 and 8, respectively. Additionally, there existed five pseudogenes. Genes including, alls, and sixs were completely lost. Sequence comparison results indicated that the highest similarity was observed betweenand, which reached 96.7%. Phylogenetic analysis revealed that the seven plants could be divided into five groups, and among them,andwere found to be gathered in the same clade, showing their highest sequence similarity.The assembly, sequence analysis, and phylogenetic analysis ofchloroplast genome provide insight into further studies on genetic structure and genetic diversity, and the results also provide evidences for evolution and phylogenetic analysis of Loranthaceae plants.
(Merr.) H. S. Kiu;(Lecomte) Danser;(DC.) Danser; chloroplast genome; sequence characterization; phylogenetic analysis
R286.2
A
0253 - 2670(2023)10 - 3273 - 08
10.7501/j.issn.0253-2670.2023.10.024
2022-10-26
台州市211人才工程经费资助(2012年度)
蒋 明,博士,教授,浙江嵊州人,研究方向为植物基因组学、植物逆境生物学及其分子调控。E-mail: jiangming1973@139.com
张慧娟,博士,副教授,浙江天台人,研究方向为植物逆境生物学及其分子调控。E-mail: zhanghj82@126.com
[责任编辑 时圣明]
