甲氨蝶呤的合成工艺改进
2023-05-18朱海溪李靖刘美朝陈彬田帅华
朱海溪,李靖*,刘美朝,陈彬,田帅华
1江苏奥赛康药业有限公司,南京 211112;2南京医科大学 药学院,南京211166
甲氨蝶呤(methotrexate,1)作为一种抗叶酸类药物,在肿瘤、类风湿性关节炎等疾病中有良好的疗效[1]。目前,文献报道用于生产甲氨蝶呤的方法主要以N-[4-(甲氨基)苯甲酰基]-L-谷氨酸-1,5-二乙酯(4)和6-溴甲基-2,4-蝶啶二胺氢溴酸盐(5)为起始原料,经取代反应得到(2S)-2-[[4-[[(2,4-二氨基蝶呤-6-基)甲基]甲氨基]苯甲酰基]氨基]戊二酸二乙酯(6),再经水解得到[2],合成路线见图1。

图1 甲氨蝶呤的合成路线
该路线操作简单、收率较高,但存在两个问题:一是5活性高(易降解)、纯度偏低(含有机杂质约8%~9%);二是在合成6的过程中,碳酸钾与氢溴酸反应生成水,蝶啶环顶端的氨基发生水解,易产生杂质C前体,并衍生为成品中的杂质C(图2)[3],难以去除(按图1 所示方法,得到6的纯度为88.58%,其中杂质C前体含量为3.58%;继续合成甲氨蝶呤,其纯度为98.84%,其中杂质C含量为0.78%)。目前,杂质C在甲氨蝶呤原料药美国药典USP40 及欧洲药典EP10.0 中的限度为≤0.5%[4,5],因此需要严格控制中间体和成品的质量。
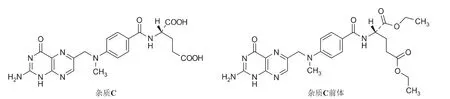
图2 杂质C 和杂质C 前体结构式
经检索[6,7],目前尚无6的纯化方法报道。本文在图1 的基础上,优化了合成路线,并开发了6的精制方法(图3)。即以4-甲氨基苯甲酸(2)、L-谷氨酸-1,5-二乙酯盐酸盐(3)为原料,经缩合反应得到N-[4-(甲氨基)苯甲酰基]-L-谷氨酸-1,5-二乙酯(4)[8],4与6-氯甲基-2,4-蝶啶二胺盐酸盐(5′)在溴化钠作用下反应得到(2S)-2-[[4-[[(2,4-二氨基蝶呤-6-基)甲基]甲氨基]苯甲酰基]氨基]戊二酸二乙酯(6)[2],6溶于N,N-二甲基乙酰胺中,搅拌下滴加对甲苯磺酸水溶液进行析晶,得到其对甲苯磺酸盐(6′),6′在氢氧化钠水溶液中脱去乙基得到甲氨蝶呤二钠(7),通过调节pH 可析出甲氨蝶呤(1)。新的合成路线能够降低6及6′中杂质C前体的含量,进而降低甲氨蝶呤中杂质C的含量。

图3 甲氨蝶呤新合成路线
2 合成实验
2.1 仪器与试剂
JY10002 电子天平(上海良平仪器仪表有限公司);DF-101s 集热式恒温加热磁力搅拌器(巩义市予华仪器有限责任公司);RAT-50D 玻璃反应釜(上海申顺生物科技有限公司);LB-450 离心机(江苏赛德力制药机械制造有限公司);FZG-15 方形真空干燥机(常州成博制药机械厂)。
4-甲氨基苯甲酸(2,南京况哲医药科技有限公司),L-谷氨酸-1,5-二乙酯盐酸盐(3,山东铂源药业有限公司),6-溴甲基-2,4-蝶啶二胺氢溴酸盐(5,上海赢瑞生物医药科技有限公司),6-氯甲基-2,4-蝶啶二胺盐酸盐(5′,广安百泉医药科技有限公司),其他试剂、溶剂均为市售分析纯。
2.2 化合物4 合成与确证
向50 L 玻璃反应釜中依次加入乙腈(8.32 kg)、N,N-二甲基甲酰胺(0.29 kg)、2(1.5 kg,9.92 mol),搅拌下降温至(5±5)℃;加入2-氯-4,6-二甲氧基-1,3,5-三嗪(1.74kg,9.92mol)和N-甲基吗啉(3.01kg,29.76 mol),(0±5)℃反应1 h 后加入3(2.38 kg,9.92 mol),室温反应4 h 后过滤,向滤液中滴加纯化水(52.5 L)析晶,过滤干燥后得白色固体4(2.68 kg,收率约80.2%),熔点(mp)88~89℃。HPLC 纯度为99.28%,ESI-MS:m/z 337.1758 [M+H]+,1H-NMR(500 MHz,DMSO-d6)δ:8.228(d,J=7.5 Hz,1H),7.678(d,J=9.0 Hz,2H),6.530(d,J=8.5 Hz,2H),6.247-6.218(m,1H),4.403-4.359(m,1H),4.114-4.02(m,4H),2.715(d,J=5.0 Hz,3H),2.430-2.400(m,2H),2.096-2.054(m,1H),2.016-1.995(m,1H),1.195-1.151(m,6H)。
2.3 杂质C 前体合成与确证
向500 mL 三颈瓶中依次加入“2.2”中4(54.6 g,162.3 mmol)、N,N-二甲基乙酰胺(270 mL),升温搅拌溶解。待溶液澄清后,加入5[69.4 g,含量78.6%(约含12%~13%左右的甲酰胺溶剂、8%~9%的有机杂质),162.3mmol],55~60℃反应2h。降温至(25±5)℃后,将反应液转移至2 L 三口瓶中,滴加1.35 L纯化水,搅拌析晶过夜。过滤干燥后得棕褐色固体66 g,即化合物6,收率约70.6%,HPLC 纯度为88.58%,杂质C前体含量3.58%(LC-MS 提取分子量512)。取出部分化合物3,经制备液相提取、纯化,得到杂质C前体1.1 g,mp 170~172℃,HPLC纯度为99.0%[色谱柱为ACE Excel 5 Super C18(250 mm×4.6 mm,5 μm);流动相A 为3.4 g·L-1无水磷酸二氢钠溶液(用1 mol·L-1氢氧化钠溶液调节pH 值至6.0)-乙腈(95∶5);流动相B 为乙腈;流速为1.0 mL·min-1;柱温为25℃;检测波长为260 nm;出峰时间为15.823 min]。ESI-MS:m/z 512.15[M+H]+;1HNMR(400 MHz,DMSO-d6)δ:11.4506(s,1H),8.4972(s,1H),8.3182(d,J=7.48Hz,1H),7.7258(d,J=8.88Hz,2H),6.8076(brs,1H),6.7964(d,J=9.00 Hz,2H),4.7948(s,2H),4.4131-4.3578(m,1H),4.1157-4.0116(m,4H),3.1981(s,3H),2.4274-2.3900(m,2H),2.1060-1.9413(m,2H),1.1936-1.1398(m,6H)。
2.4 杂质C 合成与确证
向50mL 反应瓶中加入“2.3”中杂质C前体(1g,1.95 mmol)、纯化水10 mL,搅拌下滴加10%左右的氢氧化钠(0.3 g,7.5 mmol)水溶液3 mL,滴毕后使内温在(15±5)℃反应1.5 h。过滤,向滤液中滴加39 mL丙酮,析出固体。过滤,滤饼用10 mL 纯化水溶解,5%硫酸调节pH 值至4.0~4.5,析出固体,过滤干燥后得到杂质C约0.67 g,收率约75.4%,mp 183℃~184℃,HPLC 纯度为98%[色谱柱为Waters Xbridge C18柱(250 mm×4.6 mm,5 μm);流动相A 为3.4 g·L-1无水磷酸二氢钠溶液(用1 mol·L-1氢氧化钠溶液调节pH 值至6.0)-乙腈(95∶5);流动相B 为3.4 g·L-1无水磷酸二氢钠溶液(用1 mol·L-1氢氧化钠溶液调节pH 值至6.0)-乙腈(50∶50);流速为1.0 mL·min-1;柱温为30℃;检测波长为280 nm;出峰时间为6.980 min]。ESI-MS:m/z 454.1507[M-H]-,1H-NMR(500 MHz,DMSO-d6)δ:12.283(s,1H),11.458(s,1H),8.493(s,1H),8.208(d,J=8.0 Hz,1H),7.729(d,J=8.5 Hz,2H),6.893(s,1H),6.791(d,J=9.0 Hz,2H),4.790(s,2H),4.374-4.329(m,1H),3.193(s,3H),2.325(t,J=7.5 Hz,2H),2.076-2.039(m,1H),1.941-1.894(m,1H)。
2.5 化合物6 合成与确证
向反应釜中依次加入N,N-二甲基乙酰胺(10.97 kg)、“2.2”中化合物4(2.6 kg,7.73mol),搅拌下升温,待内温升至45℃后加入溴化钠固体(1.08kg,10.55 mol)和5′(2.09 kg,7.03 mol),保温60℃反应19 h。降温至(20±5)℃后滴加纯化水(58.4 L),搅拌析晶1 h,离心干燥后得到棕褐色固体6(3.2 kg,收率约81.0%),mp 177~179℃。HPLC 纯度约93.30%,杂质C前体含量约2.41%。
2.6 化合物6 纯化条件的考察
以“2.5”中化合物6为粗品,改变良性溶剂(溶解化合物6)种类、一水合对甲苯磺酸的用量,考察杂质C前体的精制效果,结果见表1。
从表1 可以看出,使用N,N-二甲基乙酰胺/对甲苯磺酸水体系、N,N-二甲基甲酰胺/对甲苯磺酸水体系、乙醇/对甲苯磺酸水体系均可以对化合物6起到精制效果;增加一水合对甲苯磺酸的用量,可以提高杂质C前体的清除效果、提高收率,但增加到1.2 eq 后达到平台。相比于乙醇,N,N-二甲基乙酰胺、N,N-二甲基甲酰胺是更优的良性溶剂。考虑到ICH Q3C 指南中N,N-二甲基乙酰胺的毒性更小,故选择N,N-二甲基乙酰胺/对甲苯磺酸水体系、一水合对甲苯磺酸用量为6的1.2 eq 为精制条件。

表1 化合物6 纯化条件的考察
2.7 化合物6′合成与确证
按照“2.6”中优化后的条件,向500 mL 反应瓶中加入N,N-二甲基乙酰胺(100 mL)、“2.5”中化合物6(20 g,39.2 mmol),搅拌溶解。室温下滴加对甲苯磺酸水溶液[8.9 g(46.8 mmol)一水合对甲苯磺酸溶于300 mL 水中]。滴毕后搅拌析晶1 h,过滤干燥后得橙黄色固体6′(22.7 g,分子量:682.75,收率约85.0%),mp 185~186℃,HPLC 纯度96.62%,杂质C前体含量0.15%。ESI-MS:m/z 511.2387[M+H]+,1HNMR(500 MHz,DMSO-d6)δ:12.896(s,1H),9.290(s,1H),9.078(s,1H),8.728(s,1H),8.620(brs,1H),8.343(d,J=7.5 Hz,1H),7.745(d,J=8.5 Hz,2H),7.546(brs,1H),7.497(d,J=8.0 Hz,2H),7.121(d,J=7.5 Hz,2H),6.828(d,J=9.0 Hz,2H),4.880(s,2H),4.396-4.374(m,1H),4.101-4.016(m,4H),3.257(s,3H),2.405(t,J=8.0 Hz,2H),2.289(s,3H),2.099-2.060(m,1H),1.997-1.979(m,1H),1.187-1.141(m,6H)。
2.8 甲氨蝶呤二钠(7)合成与确证
依次向250 mL 反应瓶中加入纯化水98 mL、“2.6”中化合物6′(20 g,29.3 mmol),搅拌下滴加氢氧化钠(4.7 g,117.5 mmol)水溶液43 mL。滴毕后控温(15±5)℃反应1.5 h,反应结束后过滤,滤液转至1 L反应瓶中,搅拌下滴加丙酮423 mL 析晶。过滤干燥后得到黄色固体7(11.1 g,收率76.0%),HPLC 纯度为99.77%,ESI-MS:m/z 455.1763 [M-2Na+3H]+,1HNMR(500 MHz,CD3OD)δ:8.557(s,1H),7.777(t,J=7.5 Hz,2H),6.855-6.826(m,2H),4.812(s,2H),4.404-4.379(m,1H),3.209(s,3H),2.356-2.289(m,1H),2.277-2.204(m,2H),2.128-2.100(m,1H)。其中,杂质C含量约为0.07%。
2.9 甲氨蝶呤(1)合成与确证
向250 mL 反应瓶中加入“2.7”中化合物7(10 g,20.1 mmol)、纯化水100 mL,搅拌溶清后用5%硫酸溶液调节pH 至4.0~4.5,过滤干燥后得到黄色固体1(8.2 g,收率 约90%),mp 194~195℃,纯 度99.79%,杂质C含量约为0.08%[色谱柱为Waters Xbridge C18柱(250 mm×4.6 mm,5 μm);流动相A为3.4 g·L-1无水磷酸二氢钠溶液(用1 mol·L-1氢氧化钠溶液调节pH 值至6.0)-乙腈(95∶5);流动相B为3.4 g·L-1无水磷酸二氢钠溶液(用1 mol·L-1氢氧化钠溶液调节pH 值至6.0)-乙腈(50∶50);流速为1.0 mL·min-1;柱温为30℃;检测波长为280 nm]。ESI-MS:m/z 455.1791[M+H]+,1H-NMR(400 MHz,DMSO-d6)δ:8.5852(s,1H),8.1994(d,J=7.68 Hz,1H),7.7403(d,J=8.92 Hz,3H),7.5112(brs,1H),6.8327(d,J=9.04 Hz,2H),4.7934(s,2H),4.3879-4.3323(m,1H),3.2160(s,3H),2.3337(t,J=7.44 Hz,2H),2.0902-2.0429(m,1H),1.9562-1.8978(m,1H)。13C-NMR(400 MHz,DMSO-d6)δ:174.0266(4°),173.8874(4°),166.3616(4°),162.7369(4°),154.8895(4°),150.9637(4°),149.1915(3°),146.2206(4°),128.9456(3°),121.4562(4°),111.0785(3°),54.8804(2°),51.8315(3°),38.8905(1°),30.5046(2°),26.0930(2°)。
3 结论
与使用图1 所示方法得到的甲氨蝶呤(纯度98.84%,杂质C含量为0.78%)相比,使用优化后的合成路线和精制方法,6′中杂质C前体的含量由6中的2.41%(合成实验“2.5”)降低至0.15%(合成实验“2.7”),继续向后反应得到甲氨蝶呤,其纯度为99.79%,杂质C的含量为0.08%(合成实验“2.9”),符合USP40 及EP10.0 对原料药的要求。
