LC-MS/MS法测定大鼠血浆中亚胺培南浓度及其药动学研究*
2023-05-18钱敏燕杨旭萍蒋振伟左李安胡楠王莉英
钱敏燕,杨旭萍,蒋振伟,左李安,胡楠,王莉英
苏州大学第三附属医院/常州市第一人民医院 药学部,常州213003
亚胺培南(imipenem,IMP)是碳青霉烯类β-内酰胺类抗生素,对大多数革兰氏阴性、革兰氏阳性菌及厌氧菌具有抗菌活性[1],与去氢肽酶抑制剂西司他丁联合使用可防止IMP 被肾脏脱氢肽酶Ⅰ(dehydropeptidase Ⅰ,DHP-Ⅰ)代谢[2]。作为时间依赖性抗菌药,IMP 游离药物浓度超过最低抑菌浓度(minimum inhibitory concentration,MIC)的给药间隔决定了其疗效,保持适当的药物浓度高于MIC 的时间(T >MIC)可达到最佳的抗菌效果[3]。
目前测定血浆中IMP 浓度的方法包括HPLC法和液相色谱串联质谱(liquid chromatography tandem mass spectrometry,LC-MS/MS)法[4,5]。测定体内化合物时LC-MS/MS 法具有选择性好、灵敏度高以及分析时间短的优点。已有文献报道IMP 在不同种属体内的药代动力学参数[6,7],给予不同剂量的IMP会对大鼠的肾脏产生损伤[8],但目前有关不同剂量IMP 在大鼠中的药动学研究较少。本研究考察了3种不同剂量IMP 在SD 大鼠体内的药动学,为IMP相关研究提供参考依据。
1 材料与仪器
1.1 药品及试剂
注射用亚胺培南西司他丁钠(规格1 g,批号U035144,杭州默沙东有限公司),亚胺培南对照品(纯度98%,批号PNJX200214,深圳大力水手生物科技有限公司),法罗培南(faropenem,FARO)对照品(纯度80.3%,批号130532-201301,中国食品药品检定研究院);乙腈(LC 级,批号JB091230,美国Merck公司),甲酸(纯度95%,批号SHBL0331)、乙酸铵(纯度98%,批号BCCB9398)购于德国Sigma 公司。
1.2 仪器
Triple QuadTM4500MD 三重四级杆串联质谱仪,Jasper 高效液相色谱系统(美国SCIEX 公司);低温高速离心机5417R(德国Eppendorf 公司);Vortex-Genie2 涡旋震荡仪(美国Scientific Industries 公司);BT25S 电子分析天平(德国Sartorius 公司);Milli-Q 纯水仪(美国Merck 公司)。
1.3 动物
18 只雄性SD 大鼠,体重300 g 左右,由常州卡文斯实验动物有限公司提供,许可证号:SCXK(苏)2021-0013。
2 方法
2.1 色谱条件
色谱条件:Phenomenex Kinetex®HILIC 柱(50 mm × 2.1 mm,2.6 μm);流动相A:含0.1%甲酸和5 mmol·L-1乙酸铵的水溶液,流动相B:乙腈;梯度洗脱:0~0.7 min,95%B;0.7~1 min,95%→40%B;1~2.5 min,40%B;2.5~2.7 min,40%→95%B;2.7~5 min,95%B;流速:0.4 mL·min-1;进样器温度:4℃;柱温:40℃;进样体积:5 μL。
2.2 质谱条件
质谱条件:电喷雾离子化电离源(electrospray ionization,ESI),扫描方式为多重反应监测(multiple response monitoring,MRM),正离子模式;离子喷射电压:5500 V;离子源温度:500℃;雾化气(GS1):344.7 kPa;辅助气(GS2):344.7 kPa;气帘气(curtain gas):206.8 kPa;亚胺培南、内标法罗培南离子对分别为m/z→300.0/142.0,m/z→308.0/182.0;解簇电压分别为55、53 eV;碰撞能量分别为21、24 eV。
2.3 溶液配制
称取IMP 对照品适量,精密称定,甲醇定容得1.00 mg·mL-1的IMP 储备液,分装后置于-80℃冷冻保存。
称取FARO 对照品适量,精密称定,甲醇定容得0.50 mg·mL-1内标储备液,稀释得50.0 μg·mL-1的内标工作液,分装后置于-80℃冷冻保存。
2.4 标准曲线和质控样品制备
用甲醇稀释IMP 储备液得2、5、10、20、50、100、200、500、1000 和2000 μg·mL-1的标准品溶液。另稀释IMP 储备液得定量下限(lower limit of quantitation,LLOQ)质控溶液(0.2 μg·mL-1)及低、中、高浓度质控溶液(3、150、1500μg·mL-1)。取45 μL血浆,加入5 μL 上述不同浓度的IMP 标准品溶液和质控溶液,得到终浓度为0.2、0.5、1、2、5、10、20、50、100、200 μg·mL-1的系列标准血浆样品,LLOQ 浓度(0.02μg·mL-1)以及低、中、高浓度(0.3、15.0 和150.0 μg·mL-1)的质控血浆样品。
2.5 血浆样本前处理
取血浆50 μL,加入5 μL 内标FARO(50 μg·mL-1),再加入150 μL 乙腈,涡旋振荡3 min,以16 400 r·min-1离心10 min,取上清液进样分析。样本从-80℃取出到处理完成至进样过程控制在2 h 内。
2.6 方法学验证
2.6.1 专属性 空白血浆、含FARO 的空白血浆、含IMP 标准品及FARO 的空白血浆以及给药10 min后大鼠血浆样品,均按“2.5”项下方法处理后进样分析,色谱图见图1。IMP 和FARO 的保留时间分别为2.40、1.14 min,血浆中杂质和内源性物质在IMP 保留时间内无干扰,说明该方法专属性良好。

图1 血浆样品亚胺培南与法罗培南的LC-MS/MS 色谱图
2.6.2 标准曲线和定量下限 取“2.4”项下制备的系列标准血浆样品,按“2.5”项下处理后进样,建立线性回归的标准曲线。待测物与内标物峰面积比Y为纵坐标,待测物浓度X(μg·mL-1)为横坐标,使用加权(W=1/X2)最小二乘法进行线性回归运算,得直线回归方程;采用内标法对IMP 进行定量。结果表明,IMP 在0.2~200 μg·mL-1内线性关系良好,直线回归方程为Y=2.44×10-3X-1.66×10-4,r2=0.996。LLOQ 为0.2 μg·mL-1(S/N >10)。
2.6.3 准确度和精密度 取“2.4”项下制备的LLOQ血浆样品(0.02 μg·mL-1)和低、中、高(0.3、15.0 和150.0 μg·mL-1)浓度的质控血浆样品各5 份,按“2.5”项下进行处理后进样分析,连续测定3 d,计算批内(n=5)和批间(n=3)精密度,结果见表1。
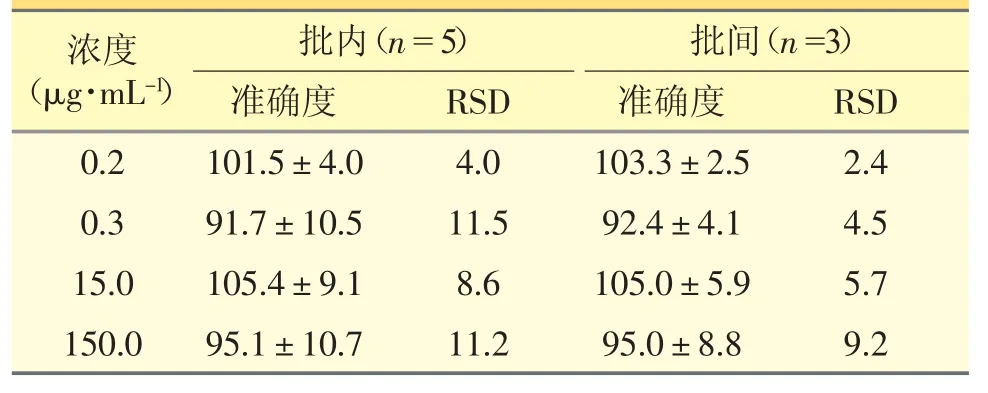
表1 亚胺培南测定方法的准确度以及精密度RSD(%)
2.6.4 基质效应和提取回收率 分别取50 μL 纯水和不同来源的空白血浆6 份,加入150 μL 乙腈沉淀,涡旋后分别加入“2.4”项下低、中、高浓度质控溶液(3、150、1500 μg·mL-1)和内标溶液,混匀后进样分析,记录纯水样本峰面积As和内标峰面积Ai之比(A),记录血浆样本峰面积A′s和内标峰面积A′i之比(B)。以峰面积比值计算基质效应,基质效应(%)=B/A×100%。
取50μL 不同来源的空白血浆6 份,加入150μL乙腈沉淀,涡旋离心后取上清,分别加入低、中、高浓度质控和内标溶液(使其浓度与低、中、高质控血浆样本浓度相同),混匀后进样分析,记录低、中、高浓度样本峰面积As和内标峰面积Ai之比(C);取“2.4”项下低、中、高浓度质控溶液(3、150、1500 μg·mL-1),按“2.5”项下处理6 份样品,进样得到低、中、高浓度样本峰面积A′s和内标峰面积A′i之比(D)。以峰面积比值计算提取回收率,提取回收率(%)=C/D×100%。
QC 样品的基质效应为91.3%~104.6%,提取回收率为88.9%~101.6%,满足生物样品的分析要求。
2.6.5 稀释可靠性 取“2.4”项下低、中、高浓度的质控血浆样品各5 份,再用空白血浆稀释2 倍(稀释后的理论质量浓度为0.15、7.5 和75 μg·mL-1)后,按“2.5”项下处理后进样以考察稀释可靠性。经稀释后低、中、高浓度的质控血浆样品的准确度分别为(101.2±4.4)%、(98.0±11.8)%和(88.3±5.2)%,RSD分别为8.7%、12.1%和3.4%。
2.6.6 稳定性 取“2.4”项下低、中、高浓度的质控血浆样品5 份,考察样品在常温2 h,4℃放置4 h、-80℃冻融3 个循环、-80℃冻存10 d 以及处理后在自动进样器中放置4 h 的稳定性,结果见表2。

表2 亚胺培南测定方法的稳定性(n=5)
2.7 药动学研究
18 只雄性SD 大鼠分为3 组,每组6 只,注射用亚胺培南西司他丁钠用适量生理盐水溶解后,分别按50、100、200 mg·kg-1的剂量腹腔注射(intraperitoneal injection,ip),并于给药后10、20、40、50、60、75、90、120、240、360 min 从眼底静脉丛采血200 μL,置于肝素化离心管中,采血后立即以3000 r·min-1离心5 min,取上清50 μL 于-80℃保存,待测。测样时将样品置于4℃冰箱中解冻,按“2.4”项下处理后进样分析。200 mg·kg-1给药的大鼠血浆样品浓度超出标准曲线线性范围,加入等量大鼠空白血浆稀释,按“2.4”项下处理后进样分析。
所得数据用Winnonlin 6.0 软件处理,采用非房室模型计算药动学参数。SD 大鼠腹腔注射50、100和200 mg·kg-1IMP 后的平均血药浓度-时间曲线见图2,主要药动学参数见表3。

图2 SD 大鼠腹腔注射IMP 后的平均血药浓度-时间曲线

表3 SD 大鼠腹腔注射IMP 后主要药动学参数(n=6)
3 讨论
反相色谱柱对极性化合物的保留能力弱,因IMP为强极性化合物在反相色谱上常出现色谱峰拖尾的问题[9]。HILIC 色谱柱所用的流动相与反相色谱柱相似,但其极性固定相对亲水化合物的保留能力强于反相色谱柱,适用于分离强极性和亲水性化合物[10]。在HILIC 色谱中乙腈分离IMP 的效果比甲醇好,选用乙腈-水作为流动相时发现IMP 的响应偏低且峰型较差,故尝试在流动相中加入酸和挥发盐。结果发现在水相加入甲酸可提高IMP 的响应,加入乙酸铵可改变峰型,因此选择0.1%甲酸和5 mmol·L-1乙酸铵作为流动相进行梯度洗脱。
IMP 在生物样本中不稳定,此外pH 值和温度对其稳定性影响较大[11];在偏碱性或酸性的条件下易生成开环降解产物;pH=1 时,高浓度IMP 还会降解生成稳定的二酮哌嗪结构化合物[12]。在IMP 检测相关文献中,常加入2-(N-吗啡啉)乙磺酸[2-(N-morpholine)ethylene sulfonic acid,MES] 或3-吗啉丙磺酸(3-morpholino propanesulfonic acid,MOPS)等非亲核试剂作为稳定剂[4,13]以提高其稳定性。但长期使用稳定剂会降低质谱灵敏度,对质谱造成损伤[14]。本文也考察了操作过程中影响IMP 稳定性的因素,不同条件下的稳定性均在可接受范围内,因此在本研究未使用稳定剂。在整个实验过程中需控制处理温度和时间以减少IMP 的降解,取血后需立即离心取上清置于-80℃中保存。另有研究显示未使用稳定剂的IMP 血浆样品(30 mg·L-1)可在室温下放置2 h、4℃下放置4 h 或-70℃下储存6个月[13]。
在临床剂量范围内IMP 的药代动力学是线性的[15],本研究发现各组大鼠的Cmax(r=0.916,P <0.01)和AUC0-t(r=0.961,P<0.01)与剂量呈线性相关。参考大鼠静脉注射(intravenous injection,iv)IMP(50 mg·kg-1)后所得AUC0-∞(7 713.89±2 608.39)μg·min·mL-1[16],结合本研究的AUC 数据,计算得到绝对生物利用度为68.2%,与IMP 肌肉注射的生物利用度(70%~80%)[17]接近。Peng L等[16]研究了SD 大鼠经静脉注射IMP(50 mg·kg-1)的药动学参数,CL(0.007±0.002)mL·min-1·g-1、Vd(0.319 ± 0.18)mL·g-1、t1/2(29.295±12.972)min 及MRT0-∞(48.457±9.18)min与本研究结果较为一致,但AUC0-∞(7 713.89 ±2 608.39)μg·min·mL-1、Cmax(180.5±64.859)μg·mL-1和tmax(2.5±1.22)min 存在差异,因为ip 给药后药物需经腹膜吸收入血后再分布至全身。与本研究结果相比,Boulamery A等[18]研究了Wistar 大鼠给予IMP(im 140 mg·kg-1)的药动学参数,AUC0-∞(177.38±24.58)μg·h·mL-1和Cmax(85.94±19.71)μg·mL-1偏低,CL/F(0.27±0.04)L·h-1、t1/2(0.92±0.24)h 和V/F(0.37±0.13)L 可能是肌肉注射使IMP 的释放延长[19]。综上所述,本文建立了一种LC-MS/MS 法测定大鼠血浆中IMP 的浓度,比较了3 种不同剂量IMP在SD 大鼠体内的药动学参数,可以为IMP 在大鼠中的相关研究提供参考依据。
