12例Dravet综合征患儿的临床特征及基因突变特点*
2023-05-17李佳豪艾戎刘维亮成善青王凌
李佳豪,艾戎*,刘维亮,成善青,王凌
(贵州医科大学附属医院 儿科,贵州 贵阳 550004)
Dravet综合征(dravet syndrome,DS)位列我国第一批罕见病目录第105号,是国际抗癫痫联盟(International League Against Epilepsy,ILAE)认定的一种罕见、严重的发育性癫痫性脑病[1-2]。DS多于婴儿期起病,发病率约为1/15 000~1/40 000[3-4],癫痫发作类型多样,包括全面强直阵挛发作(generalized tonic-clonic seizure,GTCS)、双侧阵挛性发作、局灶性发作(focal seizures,FS)、肌阵挛发作及非典型失神发作[5]。多数患儿1岁前发育正常,1岁后出现不同程度发育停滞甚至倒退,均伴随智力低下[6]。80%DS患者是由于编码电压门控钠离子通道α1亚单位(encoding the alpha 1 subnit of the sodium channel,SCN1A)基因突变引起[7],因此基因诊断对于疾病早期识别有重要意义。本研究选取儿童神经科诊治的DS患儿临床及基因突变资料进行分析,为该病的诊断和治疗提供指导。
1 对象与方法
1.1 研究对象
选择2017年1月—2022年6月儿童神经科诊治的DS患儿为研究对象,要求符合DS诊断标准[8]:(1)1岁以内常以热性惊厥为主要表现,起病高峰期为生后6个月,多表现为长时间的GTCS或半侧阵挛发作;(2)1岁后出现多种发作类型,包括肌阵挛发作、不典型失神发作及FS等;(3)发作具有显著的热敏感特征;(4)发生癫痫持续状态(status epilepticus,SE)风险较高;(5)早期发育正常,1岁后智力运动发育出现落后或倒退,可伴有共济失调和锥体束征;(6)1岁以前脑电图多为正常,1岁后可出现全导棘慢波、多棘慢波或局灶性、多灶性痫样放电;(7)抗癫痫药物(antiseizure medication,ASM)疗效不佳。共纳入DS患儿12例,男7例、女5例,截止随访年龄3~13岁、平均6岁,首次发作年龄4~15月、中位12月。本研究获医院医学伦理委员会批准(2022伦审第381号)。
1.2 研究方法
1.2.1一般临床资料及辅助检查信息 记录患儿一般临床资料,包括年龄、出生史、生长发育史、家族史、起病时间、确诊时间、癫痫发作诱因及类型、神经系统体征、量表检查、视频脑电图(video electroencepha-logram,VEEG)检测、头颅核磁共振检查(magnetic resonance imaging,MRI)、基因报告、ASM的种类及预后。
1.2.2神经发育迟滞评估 6岁及以下DS患儿采用Gesell发育量表进行发育评估,6岁以上DS患儿采用韦氏学龄儿童智力量表进行智力测验。根据美国医学遗传学学院(American College of Medical Genetics,ACMG)的定义[9],将神经发育迟滞程度分为:(1)轻度 [发育商(developmental quotient,DQ)或智商(inelligence quotient,IQ)为50~70分];(2)中度(DQ或IQ为35~50分);(3)重度(DQ或IQ为20~35分);(4)极重度(DQ或IQ为<20分)。
1.2.3药物治疗方案与疗效判断 根据DS患儿癫痫发作类型及癫痫综合征选择合适的ASM,根据疗效及时进行药物联合治疗,主要药物包括丙戊酸钠[valproat,VPA;赛诺菲制药有限公司,5~30 mg/(kg·d)]、左乙拉西坦[levetiracetam,LEV;法国NextPharma SAS公司,20~60 mg/(kg·d)]、托吡酯(topiramat,TPM;西安杨森制药有限公司,200~400 mg/d)、氯硝西泮[clonazepam,CNZ;江苏恩华药业股份有限公司,0.01~0.2 mg/(kg·d)]、拉莫三嗪(lamotrigine,LTG;美国GlaxoSmithKline Pharmaceuticals公司,25~200 mg/d)及奥卡西平[oxcarbazepine,OXC;Novartis Farma S.P.A,8~30 mg/(kg·d)]。ASM治疗效果按以下标准评价[10]:癫痫无发作且临床症状消失为完全控制,癫痫发作次数较治疗前降低75%以上且临床症状得到控制为显效,癫痫发作次数较治疗前下降50%~75%且临床症状缓解为有效,癫痫发作次数较治疗前下降50%以下且临床症状无明显改善为无效;将完全控制或显效定义为疗效显著,有效或无效定义为疗效不佳。此外,DS患儿难治性癫痫诊断参考2010年ILAE标准[11]:根据发作类型,合理选择并正确使用(单用或联用均可)至少2种耐受性好的ASM后,患者无发作持续时间未达到治疗前最长发作间隔的3倍或1年(取决于两者之间何者更长)。
1.2.4基因检测方法 采集DS患儿及其父母静脉血各4 mL,置于含乙二胺四乙酸(ethylenediamine tetraacetic acid,EDTA)抗凝试管中,采用Qiagen FlexiGene DNA Kit试剂盒(德国Qiagen公司)提取基因组DNA,并对其进行质控;采用Qubit仪器精确定量DNA浓度,经Covaris破碎仪将DNA打断成180~280 bp片段,片段两端分别连接上接头制备DNA文库,使用Qubit 2.0进行初步定量,使用Agilent 2200检测文库的insert size,用荧光定量多聚核苷酸链式反应(quantitative polymerase chain reaction,qPCR)定量文库的有效浓度;运用NovaSeq 6000平台进行测序,获得fastq格式的数据;采用Burrows-Wheeler-Alignment(BWA)软件包(v0.7.15)将测序read比对到人类参考基因组(hg19版),并对结果进行质量控制,分析可能存在的拷贝数变异,进行基因相关注释及蛋白损伤分析,结合临床症状、基因遗传方式、变异频率及测序深度等相关信息选出需要一代验证的突变位点并按照ACMG遗传变异分类标准与指南进行致病性注释分析;对于疑似位点,对DS患儿及父母采用Sanger测序进行验证,通过一代测序验证,排除二代测序中假阳性的位点。
1.2.5随访及信息收集 DS患儿随访通过门诊复诊、电话、微信回访等多种形式收集用药情况、癫痫发作频率、精神运动发育情况、复查VEEG及头颅影像学检查等;随访10个月~5年,至末次随访时,1例失访,1例病故。
1.3 统计学分析
采用SPSS26.0软件对数据进行处理。满足偏态分布的计量资料采用中位数(四分位数间距)[M(P25,P75)]描述,计数资料采用例数和百分数(%)描述,组间分类变量比较采用卡方检验或Fisher精确概率法;P<0.05为差异有统计学意义。
2 结果
2.1 一般临床资料
婴儿期起病9例(75.0%),确诊年龄范围为5月~4岁、平均2(1,3)岁,因临床表现不典型或误诊为热性惊厥导致诊断延迟8例(66.7%);发作诱因为热水浴2例次、感染发热10例次、接种疫苗3例次,发作具有热敏感特点10例(83.3%);首次发作类型为GTCS发作8例(66.7%)和FS 4例(33.3%),单一发作类型6例(50.0%,GTCS 4例、FS 2例,其中1岁后出现眼睑肌阵挛1例),2种发作类型6例(50.0%,为GTCS+FS,其中半侧阵挛发作2例);发生SE 8例(66.7%);12例患儿(100.0%)病前发育均正常,截止随访时出现不同程度的发育迟缓,但治疗后有进步,查体无异常神经系统体征;全面发育迟缓3例(25.0%,其中共济失调2例),运动发育迟缓2例(16.7%),语言发育迟缓7例(58.3%);12例(100.0%)无异常出生史,有癫痫或热性惊厥家族史3例。
2.2 辅助检查
DS患儿VEEG背景慢化11例(91.7%);局灶性癫痫样放电9例(75.0%),放电位置多样,多位于一侧或双侧颞区,其次为顶区和后头部;起病初期未见明显异常3例(25.0%);1例在监测中见可疑右侧顶、顶中线区起始FS 2次;头颅MRI示双侧海马硬化1例,双侧颞角扩大1例,颞部蛛网膜囊肿1例,无异常9例。
2.3 基因检测
12例患儿检出SCN1A基因突变9例[75.0%,其中De novo7例(77.8%)],变异类型为错义变异5例(55.6%)和移码变异4例(44.4%);检出编码电压门控钠离子通道α9亚单位(encoding the alpha 9 subunit of the sodium channel,SCN9A)基因错义变异1例(8.3%),均来源于父亲;检出SET含结构域蛋白2(set domain-containning protein2,SETD2)基因突变和神经元烟碱胆碱能受体α多肽2(cholinergic receptor neuronal nicotinic alpha polypeptide2,CHRNA2)基因突变各1例(8.3%),均来自于父亲。见表1。
2.4 治疗方案及效果
12例患儿均选用多种ASM治疗(其中二联4例、三联5例及四联3例),符合难治性癫痫6例,VPA12例次、LEV8例次、TPM9例次、CNZ2例次、OXC3例次(均已减停)及LTG1例次(已减停),药物有效率为75.0%,随访结束时发育迟滞评估程度为轻度5例、中度3例及重度4例。见表1。
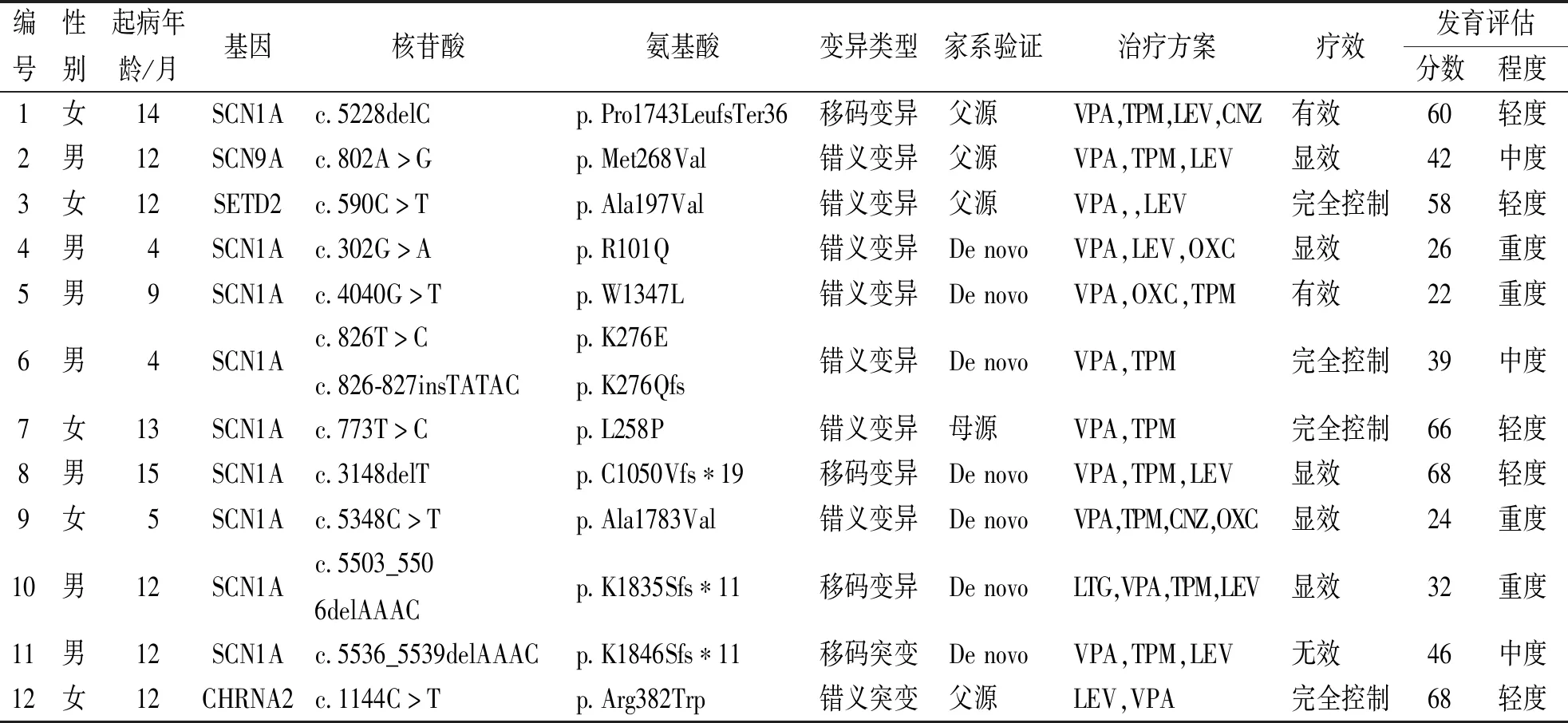
表1 12例DS患儿基因突变、治疗及预后Tab.1 Gene mutation,treatment,and prognosis of 12 children with DS
2.5 SCN1A基因突变与癫痫持续持续状态的关系
SCN1A基因突变(SCN1A+)与无SCN1A基因突变(SCN1A-)患儿SE、精神发育情况比较,差异无统计学意义(P>0.05,表2)。
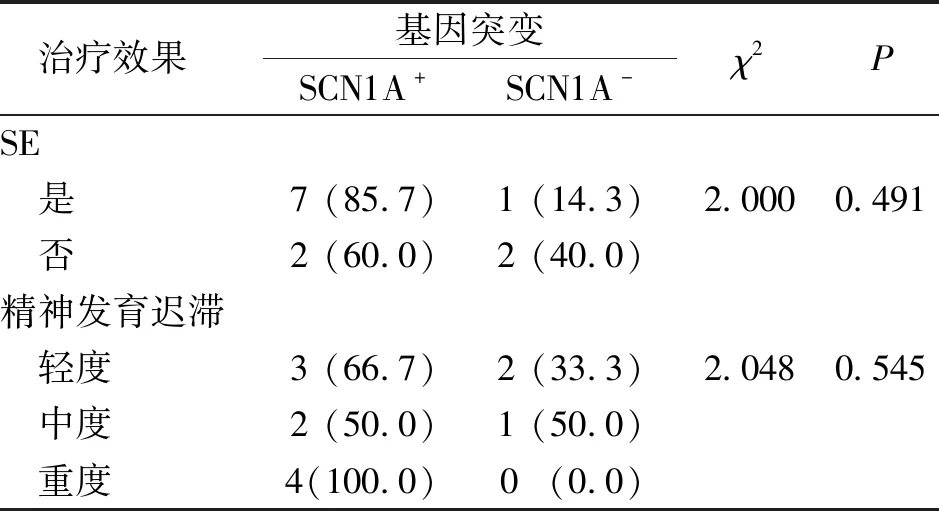
表2 DS患儿中SCN1A基因突变与SE、精神发育的关系[n(%)]Tab.2 Association of SCN1A gene mutations with SE and mental development in children with DS [n(%)]
3 讨论
DS是一种与遗传相关的难治性癫痫综合征,75%患儿有SE[12]。本研究中66.7%患儿有SE,低于文献报道。患儿通常会于发热或接种疫苗后出现抽搐发作[13],因此在发病初期该病易被误诊为热性惊厥,导致诊断延迟。本研究中66.7%患儿出现诊断延迟。He等[14]研究表明,DS最常见的首次发作类型是GTCS(52.0%),其次是半侧阵挛发作(35.0%),而其他发作类型如非典型失神发作、肌阵挛发作等通常出现于1~5岁。本研究中66.7%DS患儿首次发作类型为GTCS,发生率略高于上述研究;有1例1岁后出现眼睑肌阵挛,与该文献相符。因此,早期临床及基因结合诊断在DS诊疗中占重要地位。
DS初期VEEG可为正常,易被误诊为热性惊厥;1岁以后超过90%患儿脑电图异常,局灶性癫痫样放电或弥漫性背景慢化的可能性随着年龄的增长而增加[15]。本研究中91.7%患儿VEEG出现背景慢化,25.0%患儿病初VEEG未见明显异常,后随着病情进展出现不同程度的背景慢化及痫样放电,75.0%患儿VEEG见局灶性癫痫样放电及放电位置多样(多位于一侧或双侧颞区,其次为顶区和后头部)。这提示有DS相关临床表现的患儿需定期监测脑电图,以便早期监测到痫样放电。
DS突变约10%与父母遗传有关,且父母表型轻微或正常,有些仅表现为热性惊厥[16]。本研究DS突变来源于父母的2例SCN1A突变患儿中,编号1患儿基因突变来源于父亲,其父幼时有热性惊厥,现无发作;编号7患儿基因突变来源于母亲,其母幼时有热性惊厥,产后强直阵挛发作2次,这与文献描述相符。7例DS患儿为De novo,其中突变位点无文献描述6例,有SE 5例;7例均使用2种及以上ASM治疗,疗效不佳2例、疗效显著5例,提示新发突变致病意义更大,更易出现SE。SCN1A变异类型很广,包括无义变异、错义变异、剪接变异、移码变异及缺失重排等[17]。本研究有9例SCN1A基因变异DS患儿中,错义变异占55.5%,发生率较文献报道高[18]。Ragona等[19]研究表明,6岁以后的DS患儿均有智力发育落后,但归因于SCN1A基因突变还是多种因素共同作用的结果尚无定论。本研究中SCN1A+与SCN1A-患儿在是否出现SE及精神发育迟滞严重程度上无差异,提示基因突变种类不同与DS的临床表现严重程度及疗效关系并不密切,这与李丹等[20]研究结果相符。
除SCN1A外,其他基因如SCN2A、SCN8A、SCN9A、原钙蛋白19(protocadherin19,PCDH19)、编码电压门控钠离子通道β1亚单位(encoding the beta 1 subunit of the sodium channel,SCN1B)等基因突变也可以参与DS或DS样表型[21];其中SCN9A突变被认为与发热性癫痫有关,且可能与SCN1A共同导致DS[22]。本研究结果显示,SCN9A、SETD2及CHRNA2基因突变各1例,SETD2为常染色体显性突变,该突变不仅与亨廷顿病有关,也可导致轻度整体发育迟缓、中度受损智力残疾伴言语困难和行为异常[23]。编号4患儿的SETD2基因突变来源于父亲,患儿及其父亲、姐妹均为杂合突变,且都有癫痫发作、精神发育迟滞,考虑该基因为致病基因。CHRNA2为常染色体隐性遗传,其突变导致夜间额叶癫痫4型及良性家族性婴儿6型[24];编号12患儿及其父亲均为杂合突变,但其父亲无癫痫发作,考虑为该致病基因外显不全或该突变不致病。这2种基因目前无研究表明与DS有明显的联系,但患儿临床表现、发育情况及脑电图均符合DS诊断标准,故予以纳入。
多数DS患儿需多种ASM联合治疗,以期控制癫痫发作,但仍可会出现耐药性癫痫[25]。首选ASM为VPA、氯巴占,其次为TPM、唑尼沙胺、司替戊醇及吡仑帕奈。氯巴占、司替戊醇等药物在中国大陆地区未广泛使用,对患者的治疗造成困难,且LTG、卡马西平和OXC等避免在DS中使用[26],因此传统ASM仍是治疗首选。本研究患儿均尝试多种ASM治疗,疗效显著10例,提示大多数DS患儿的癫痫发作可控制;有4例患儿病初予OXC、LTG后发作得以短暂控制,经基因检查明确诊断后逐渐减停,未再发作,提示尽早完善基因诊断有利于指导药物治疗。本研究9例SCN1A+患儿治疗效果为疗效显著6例、疗效不佳2例;精神发育迟滞程度为3例轻度、2例中度、4例重度;3例SCN1A-疗效均为疗效显著,精神发育迟滞程度均为轻度,上述对比表明DS中SCN1A-突变预后较SCN1A+好,因此,基因检测有助于DS预后评估。
总之,DS诊断已转向基因精准诊断,对于起病早且发作频繁的发热伴惊厥患儿,应警惕DS可能,尽早行遗传学诊断,有助于早期确诊、指导ASM选择,给予个体化的治疗,最大限度改善患儿的远期预后。
