In vivo Continuous Evolution*
——An Evolving Tale From Phage Towards Eukaryotic Genome
2023-05-16JIAWenPENGYing
JIA Wen, PENG Ying
(BGI College & Henan Institute of Medical and Pharmaceutical Sciences, Zhengzhou University, Zhengzhou 450052, China)
Abstract Evolution is the natural history where biological diversity is generated and retained. To expedite the evolution process in the lab to achieve specific functional optimization by artificial selection, targeted mutagenesis boosts are introduced into fastproliferating prokaryotes and simple eukaryotes. If tolerated, the functional diversity a fast-evolving system generated offers unique opportunities to obtain artificially engineered bio-molecules best suited for bioengineering applications. In this perspective, we discuss the current state of art in vivo continuous evolution platforms, focusing on advances made in both phage and yeast artificial evolution. Their successful applications in biotechnology are presented, followed by a brief outlook of near-future developments in this burgeoning field.
Key words in vivo evolution, directed evolution, phage-assisted-continuous evolution, protein engineering, targeted mutagenesis
The authors dedicate this article in memory of Dr. Chen-Lu Tsou (1923-2006), a great protein biochemist, a generous mentor and a dedicated truth fighter. The first but most valuable life advice one of the authors (PENG Y)learned, from an early correspondence with Dr. Tsou in the pre-internet era, is from Isaac Newton’s famous “If I have seen further than others, it is by standing upon the shoulders of giants” quote. Life by itself, indeed never stops evolving itself in all possible forms. The pursuit towards the beauty that truth holds, usually with the key insights gained by dissecting its most fundamental molecular components, has inspired generations of youth turned into giants themselves, by first climbing onto the shoulders of Dr. Tsou. The thrill of the climbs for sure is vast, while the ebb of conquering shoulders by shoulders will never retreat.
Directed evolutionin vitrohas been best demonstrated by the success of systematic evolution of ligands by exponential enrichment (SELEX) in making nucleic acid aptamers, binding to target ligands with high binding affinity and specificity[3].SELEX is based on a very large oligonucleotide library that is obtainedviachemical synthesis on demand, which can be easily amplified exponentiallyin vitro, with the possibility of introducing further random sequence drifts throughin vitroamplification.The ability for nuclei acid to be exponentially amplified directly is the reason why SELEX approach is mainly used for nuclei acid-based binders. Proteins,on the other hand, cannot be readily amplified in a molecule-to-molecule manner. Therefore, the information a given protein molecule is carrying has to be associated with a fast amplifiable intermediate.In the end, selection based on the special property of any target protein need to retain an integral unit pairing the protein of interest and its information carrier. Further amplification of the carrier, with the capability of introducing further diversification drifts during its amplification, enables continuous expansion of the potential candidate pool for functional exploration. Since coupling a protein to its coding nucleic acid is difficulty to achieve and impossible to decode autonomouslyin vitro, such need pushes for the development of variousin vivoevolution schemes,using intact cells or viruses as the information carrier for the protein of interest under selection. In the recent decade,in vivodirected evolution has become a highly active research and development arena for protein biotechnology. This strategy carries additional advantages of avoiding artifacts introduced by human interventions, reducing significant large manual workload as well as the time of human consumption[4].
Among variousin vivocontinuous evolution schemes, the phage and yeast systems are at the chassis of current research spotlights[5]. Therefore, we focus our discussions on directed evolution approaches developed in those systems.
1 Phage
Among the virus-basedin vivoevolution systems, the phage-assisted continuous evolution(PACE) system is the most mature one. The life cycle of phage is short, for example, the M13 phage used in PACE has a life cycle of only 10 min. By linking the proliferation of bacteriophages to the activity of the protein of interest (POI), rapid and continuous evolution of POIs can be achieved in cycles of short duration. As anin vivoevolutionary system, the PACE system does not require the creation of molecular diversity libraries artificially, and has a higher evolving rate than mostin vitroevolutionary systems,since phage genome can tolerate much higher mutation rate[4,6].
1.1 PACE
The phage's coat protein pIII is encoded by the gIII in M13 genome, which is key to its maturation and infection. To complete directed evolution without constant human interference, PACE uses a modified version of the M13 phage, called selection phage(SP), to continuously infect the hostE. coli. In selective phage genome, its gIII is replaced by the gene of interest (GOI). Therefore, due to the inability of making pIII, SP cannot produce infectious phage particles on their own, and will express a protein of interest instead to fulfill a particular task in hostE. coli. As a supplement, gIII is conditionally provided by a separate accessory plasmid (AP) of the hostE. coli,as part of a selection system that activates the expression of pIII strictly upon the desired activity of the POI. In combination, SP can only be propagated when sufficient pIII is generated through optimizing POI in term of controlling gIII expression(Figure 1a). PACE uses DNA polymerase with a high mutation rate to boost the error rate during phage replication. This is achieved by including mutagenesis plasmid (MP) in hostE. coli. On MP, the dominant negative variants of theE. coliDNA Pol III proofreading domain increases the mutation rate to more than 300 000 times above the basal level,allowing steady accumulation of sufficient number of mutations in SP genome. After the introduction of mutations by MP-expressed error-prone DNA polymerase, the SP carrying the mutant protein with enhanced activity will have significant proliferative advantages over other SPs due to the increase in viral infectivity from pIII production. Therefore, the successful proliferation of SP is associated with the activity of POI. Over many generations through PACE evolution, the number of SPs containing the preferred protein coding sequence (CDS) will be overwhelming among surviving SPs[7-9](Figure 1a).
The core of PACE technology is the continuous evolution of SP, which can be carried out in a continuous flow Chemostat-Lagoon pump system.The simplest PACE system consists of just two flasks,one as Chemostat and one as lagoon (Figure 1a). In Chemostat, the hostE. coliculture is continuously pumped into Chemostat, while the excess culture and cells are transported to the waste tank, allowing the cells in Chemostat to maintain a constant volume under optimal healthy growth conditions. The lagoon is filled with hostE. colicultures together with pool of selection phages. Importantly, the lagoon is constantly infused with fresh, uninfectedE. colifor infection by SPs, while the infected cells in the lagoon are constantly excreted as waste and the volume of the lagoon remains constant throughout the experiment(Figure 1a). Initially, a SP library carrying a single sequence species or a limited sequence pool can be used to infect of the hostE. coliculture without selection. At the same time, upon induction in the lagoon, gene expression on MP within host cells leads to elevated mutations in the phage genome. If mutations in the GOI on the phage module can in turn trigger the production of pIII proteins, infective daughter phages can be generated. These newly produced daughter phage strains can better expand themselves through infecting fresh hostE. colicell infused into the lagoon (Figure 1a). At the same time,most phage modules cannot initiate gIII expression in host cell, therefore, their infection cycle will abort quickly. Upon continuous outflow from the lagoon,the concentration of those incapable SP in the lagoon will plunge to near zero quickly. Therefore, by building such a system and applying appropriate flowing rate matching the speed of phage evolution and propagation, the PACE system allows selected phages to reproduce continuously in the lagoon for hundreds of generations without external interference(Figure 1a). The key to build a successful PACE system is devising a rational scheme coupling the desired property of POI to pIII production. Applying the appropriate selection pressure by adjusting the flow rate in and out of lagoon can be at times challenging, since empirical rate of continuous evolution and fitness are hard to measure quantities before experimentation[7].
1.2 PANCE
Based on PACE, established a phage-assisted non-continuous evolution (PANCE) technique[10].Unlike PACE's continuous strategy, PANCE harvests and screens one round every six hours by adding phages manually for further mutations[11](Figure 1b).This discontinuous system can be performed directly on the petri dish, reducing the strict dependence on experimental instrumentation such as bioreactors[7].With the cost of more manual involvement, PANCE offers higher flexibility to adjust the selection pressure as well as temporal duration of any givenin vivoevolution selection experiment.
1.3 IDE
PACE single-pass evolutionary DNA sequences have a limited length (typically less than 5 kb due to the limit of M13 phage genome packaging), making it difficult to screen for complex phenotypes. Al'abriet al.[12]have recently developed a method for largescale DNA directed evolution using P1 phages instead of M13, named inducible directed evolution (IDE).This method uses three plasmids: P1 contains the P1 phage fractional sequence (the DNA to be mutated);mutator plasmids carries inducible expression ofdanQ926,dam,seqA,emrR,ugiandcda1, which collectively reduce the efficacy of the DNA repair system and promote the mutation frequency; phage plasmid includes the lysis control module of the mild phage, to maneuver phage lysis behavior. IDE enhances the ability and utility of a complex directed evolution approach inE. coliwith a large phage genome.

Fig. 1 Schematic diagrams of phage-assisted continuous evolution(PACE) and related variants
1.4 PRANCE
In order to achieve high throughput and precise control of environmental factors in the evolutionary process, Debenedictiset al.[13]developed a robotbased directed evolution platform based on PACE,named PRANCE (phage-and robotics-assisted nearcontinuous evolution). This improvement has important implications for the automated development of continuous directed evolution, and creates conditions for studying the influence of various environmental variables on directed evolution.
The poor betrothed19 bride dressed herself entirely20 in black,25 and when she thought of her future bridegroom, tears came into her eyes. Nothing but contempt and mockery fell to her lot from her sisters. Take care, said the eldest, if thou givest him thy hand, he will strike his claws into it. Beware! said the second. Bears like sweet things, and if he takes a fancy to thee, he will eat thee up. Thou must always do as he likes, began the elder again, or else he will growl7. And the second continued, But the wedding will be a merry one, for bears dance well. 26 The bride was silent, and did not let them vex21 her. Bearskin, however, travelled about the world from one place to another, did good where he was able,27 and gave generously to the poor that they might pray for him.
1.5 SPACE
Although PACE technology can achieve dozens of rounds of directed evolution in a single day with continuous flow with bioreactors, evolving multiple target proteins simultaneously with PACE is unrealistic since the limited availability of sophistic pumping and bioreactor setups. Recently, Weiet al.[14]have developed a spatial phage-assisted continuous directed evolution system (SPACE), which combines the effect of bacterial spatial migration movement,and phage infection, and replaces the continuous culture device required for continuous phage expansion in the PACE system with semi-solid agar medium (Figure 1c). SPACE greatly simplifies the system setup, with the additional advantages of direct visualization of evolution and the capability for simultaneous large-scale parallel evolutionary implementations.
2 Yeast
The PACE system and its improved version as summarized above are based on the phage directed evolution system. While the choice with phage is simple since its fast proliferation speed and robust tolerance to high mutation rate in its genome.However, phage based directed evolution comes with its natural limitation, such that its evolution process is nonautonomous since the completion a phage lytic cycle is always dependent on availability of the host.To achieve a simpler, autonomous directed evolution in an eukaryotic cell, yeast was picked up as a first model of choice due to its ease of culture and fast proliferation rate. However, like most eukaryotes,high mutation rate is accompanied by overwhelming DNA damage response, which is extremely detrimental to cell survival if mutagenesis is boosted over a threshold level in the genome scale[15].Therefore, such obstacle has to be first overcome by introducing high frequency mutation on the gene of interest without compromising the rest of genome.
2.1 OrthoRep
OrthoRep, is a first successful example for such innovative approach, taking advantage of a naturally occurring orthogonal replication system in yeast.Similar to the PACE system, OrthoRep uses a faulty DNA polymerase to introduce mutations during GOI replication. OrthoRep's ingenuity lies in the fact that it bypasses the limitations of mismatch extinction through an orthogonal replication system exclusive in the cytoplasm such that the nuclear genome is spared of the error prone replication (Figure 2). Such setup is based on the “killer” plasmid system identified in some natural fungal species[16-17]. The “killer”plasmids comprise of two cytosolic linear DNA,whose DNA replication relies of protein based priming and dedicated DNA polymerases encoded on the plasmids themselves. Specifically, sequences on p1 plasmid of the “killer” plasmid set are dispensable for the autonomy of the “killer” set, while its dedicated replication machinery DNAP1 can function properly if encoded by a nuclear plasmid[18]. Therefore in the current OrthoRep system, the GOI can be placed on the p1 plasmid, while the original p2 plasmid is left intact, providing other essential components for this orthogonal replication system. If all original p1 plasmid is replaced by the engineered ones without DNAP1 sequence, mutagenesis rate can be specifically boosted by ~1×10-5substitutions per base through providing the yeast with an error prone DNAP1 encoded on a nuclear plasmid[18-20](Figure 2).
2.2 EvolvR
Unlike several previously described systems,EvolvR is a CRISPR-Cas9-based targeted mutation method developed by Dueber Lab at UC Berkeley,providing a new way to generate site specific genetic mutants.

Fig. 2 Schematic diagram of OrthoRep continuous evolution system in yeast
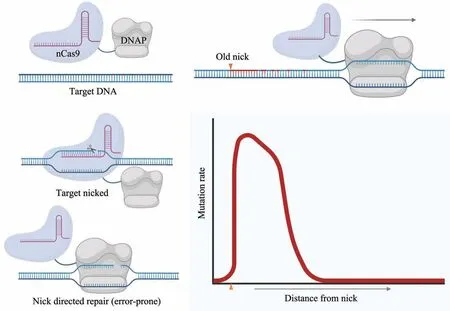
Fig. 3 Schematic diagram of EvolvR continuous evolution system
Similar to error-prone PCR, EvolvR uses errorprone polymerases to introduce mutations. To achieve the site specificity of error introduction, the errorprone PolI3M DNA polymerase is fused with nicking Cas9 (nCas9). Therefore, by using nCas9 to direct PolI3m to specific genomic loci, EvolvR allows targeted mutations at sites of interest through PolI3m mediated DNA synthesis on a nicked DNA strand(Figure 3). gRNA is used to direct the PolI3M-nCas9 complex to the site of interest in the genome (Figure 3).If a nick is created locally, PolI3M then binds to the incised DNA and introduces mutations extending from the free 3' end from the nick, while its native endonuclease activity degrades the original, displaced strand during nicked strand extension (Figure 3).EvolvR allows continuous diversification of all nucleotides at specific sites. Its modular nature provides the versatility and flexibility to adjust systematic mutation rates and editing windows[21].Due to the cross-species feasibility of CRISPR,EvolvR was also successfully implemented in yeast systems, called the yEvolvR system[22], and in theory any organism with feasible genetic transformation tools, However, the PolI3M DNA polymerase is significantly more likely to synthesize error containing DNA strand near the nicking site,therefore, the frequency of mutagenesis will drop quickly at a given distance away from the nick[23](Figure 3). This uneven mutagenic property therefore makes EvolvR most useful only when local sequence changes are required in a focal manner.
2.3 ICE
In addition to the use of error-prone DNA polymerases, the low fidelity of the reverse transcription process is also been used for mutation generation duringin vivocontinuous evolution. Crooket al.[24]used the reverse transcription transposon Ty1 to establish a continuously evolved system ICE (in vivocontinuous evolution) in yeast (Figure 4). Ty1 is a naturally occurring long-end repeat reverse transcription transposon in yeast, which first makes a RNA intermediate during the transposition process,followed by transposition with reversed transcript cDNA. A galactose-induced promoter (GAL1p) is added to the front end of the Ty1 transposon to induce the transcription of the transposon; the target gene to be evolved is introduced between 5'-LTR and 3'-LTR,in a direction opposite to the transcription direction of the transposon (Figure 4). In the presence of galactose,the transposon initiates transcription and excises a synthetic intron, but the GOI is not translated because of the orientation of the primary transcript make it noncoding. The mRNA is then reverse-transcribed into cDNA, reintegrated into the genome, and finally the protein of interest can be expressed. The errorprone reverse transcription process enabling the continuous transposition of Ty1 pushes for a continuous evolution of the POI it carries (Figure 4).With the screening pressure for a specific protein trait,directional evolution can be achieved for key players on a given metabolic pathway. However, with ICE mutation system, the transposition frequency directly affects the frequency of mutagenesis. In addition,independent transpositions coexist in the genome of the selected cell, complicating how to discern which evolved protein variant leads to the particular desired cellular phenotype. These complicating factors are likely to affect its applications in continuous evolution.

Fig. 4 Schematic diagram of ICE continuous evolution system in yeast
2.4 TRIDENT
Currentin vivomutagenesis techniques have multiple restrictive requirements such as low spontaneous mutagenesis rate, inability to modulate mutation profiles, and the need to use specific host/virus pairings and orthogonal replication systems.Recently, Cravenset al.[25]have developed a T7 RNA polymerase-guidedin vivotargeted mutation method:targetedin vivodiversification ENabled by T7 RNAP(TRIDENT). TRIDENT provides a mutation rate of more than 10-3substitutions per basein vivo, with >1 000-fold on-target mutagenesis specificity, and mutational diversity for all four DNA nucleotides can be adjusted in a 100-fold range. The core rational of TRIDENT is to fuse T7 RNA polymerase based transcription with mutagenesis, which uses T7 RNA polymerase for targeted transcription and recruit mutagenic enzymes on the transcript gene locus.TRIDENT consists of three components: (1) C∶G>T∶A substituted cytosine deaminase; (2) A∶T>G∶C substitution of engineered adenosine deaminase;(3) DNA repair factors that further enhance the diversity of A/T/G mutations. A crucial limitation of the TRIDENT system is that this approach is only feasible for foreign transgene driven by a T7 promoter. All endogenous loci can not be targeted due to their inaccessibility to T7 RNA polymerase.
3 Applications
3.1 Base editor
Single-base editing uses a Cas9 nickase to open a single-stranded DNA loop at a specific genomic site,and the bases in the editing window (usually a few nucleotides) in this region can be modified by a fused nucleotide deamylase to achieve base switching.Current single-base editing techniques include two main categories: cytidine base editors (CBEs), which can convert C/G to T/A base pairing, and adenine base editors (ABEs), which convert A/T to G/C pairing[26].Gene editing with base editing offers better sequence conversion precision comparing to Cas9 nuclease, but there are also limitations such as byproduct editing and limited recognition sites, which is of great significance for further improvements of base editors[27].
3.1.1BE-PACE
Recently, Thuronyiet al.[28]have developed a phage-assisted continuous evolution single-base editing system (BE-PACE) based on the PACE system, which can greatly improve the editing efficiency and target sequence compatibility of CBE,with the selection pressure applied by a specific on demand guideline. The researchers used the BE-PACE system to overcome the shortcomings of the target sequence limitation of traditional CBE and evolved three types of cytosine single-base editors, which were confirmed byin vitroluciferase reporter system and mammalian cells that the evolved CBE system has an increased base editing performance and expanded editing range. When BE is applied to base precision editing, there are often problems with undesired bystander mutations arisen from off-target editing. Recently, Chenet al.[29]introduced two mutations, L145T and N108Q, to TadA-8e deaminase in ABE8e through rational design, and developed accurate and safe ABE9, successfully retaining high editing activity while significantly narrowing the editing window and eliminating the editing activity of cytosine. Subsequent mutation and screening of BE by PACE is expected to obtain more diverse BE tools that can be used for base-precise editing more quickly.
3.1.2Mitochondrial base editing
Development of gene editing tools for mtDNA has been a long-term goal in the field of mitochondrial genetics. Previously, Toddet al.[30]discovered a bacterial toxin, DddA, that can catalyze the conversion of cytosine to uracil on double stranded DNA substrates. Splitting DddA into two halves abolishes its endogenous toxicity associated with its nonspecific, uncontrolled mutagenic capability. At the same time, reconstituting a functional DddA deamylase can be precisely controlled by fusing its each half with transcriptional activator-like effector array proteins (TALE) targeting adjacent sequence on mitochondrial genome.Therefore, an RNA-free DddA-derived cytosine base editor, DdCBE, catalyzes the conversion of C/G to T/A in mtDNA. Recently, David Liu's team[31]has carried out targeted modification of DdCBE through PACE and PANCE technology, and developed two upgraded versions of DdCBE, which not only have higher editing efficiency, but also have a wider range of editing. This study provides a new tool for efficient gene editing in mitochondria as well as nuclear genome.
3.2 Antibody
Morrisonet al.[32]have used upgraded PACE to evolve single-stranded variable region antibody fragments (scFv) that are misfolded and aggregated in the cytoplasm ofE. coli. Much improved variant with 5-fold increase of expression and simultaneous retention of binding activity was obtained in only 3 d.Furthermore, variant with enhanced stability without disulfide bonds was evolved from original scFvs in only 5 d. Recently, Wellneret al.[33]established the autonomous hypermutation yeast surface display(AHEAD) system, by combining OrthoRep with yeast surface display systems. They inserted the antibody fragment into the p1 killer plasmid, and realized antibody hypermutation in yeast cells through spontaneous super mutation of yeast. At present, the AHEAD system lacks a mutation control switch,therefore the mutagenesis process will persist forever.A more effective antibody discovery platform will prefer the mutagenesis process turned off once desirable hits are established[34].
3.3 Enzyme
Since 2011, Liu's group[8,35-37]has successfully engineered a variety of enzymes using PACE, such as T7 polymerase, TALEN, spCas9 variants. Using the recently developed SPACE system, Weiet al.[14]paralleled the ability of T7 RNA polymerase to recognize randomized promoter sequences, resulting in a library of promoter variants-RNA polymerase mutations that can be used for further modification and engineering applications. With the ICE system,Crooket al.[24]evolved URA3 to obtain mutants with increased catalytic activity, and successfully evolved polygenic metabolic pathways to improve the xylose metabolism capacity of yeast.
3.4 Drug resistance
There have been a few reports on the use ofin vivoevolution system in drug resistance research[19,38].By encoding plasmodium falciparum dihydrofolate reductase (PfDHFR) on OrthoRep, Ravikumaret al.[19]were able to evolve inhibitor resistant PfDHFR in 90 independent replicates after a simple serial passage of cells in the presence of the PfDHFR inhibitor pyrimethamine. Similarly, Cravenset al.[25]have used TRIDENT to achieve saturating mutagenesis for pyrimethamine resistance. Both studies revealed the convergence of all replicates on a few shared coding mutations on PfDHFR. Dueber's lab[22]used EvolvR to discover new genotypes that cause spectinomycin resistance in bacteria. While there are many known spectinomycin resistant mutations, several new mutations were identified through targeting EvolvR to five dispersed regions of the spectinomycin target generpsE. These results not only reveal potential drug resistance genotypes, but also provide a better understanding of key residues of this protein-drug interaction.
4 Perspective
4.1 In vivo continuous evolution feeding computational protein design
The greatest challenge for protein design is the vast possible sequence space available to explore any given design task, even for a small or moderate sized single subunit protein. Modern deep learning has greatly facilitated the reliability of machine-based learning, although most efficient deep learning models requires large “training” datasets[39], which provides a comprehensive “cost” function estimate regarding a given biochemical function or protein property. Such data is sparse in term of biological molecules, with the further complication that most natural proteins are“optimized” through long evolution process to fit its native functional niche. Continuousin vivoevolution provides a fast and feasible mean to generate unbiased“fitness” measurement, with quantitative power to explore the local “energy landscape” with unbiased,continuous introduction of protein sequence variations. A cell-by-cell quantitative measurement of phenotype score, in combination of retrieving sequence information of POI can be accelerated by single cell based sequencing technology, following cell-by-cell quantification and barcoding through microfluidics platforms or in-situ sequencing[40].
We envision combining the power of autonomous directed evolution with deep learning will move forward the realm of protein design from a structure-based domain, to various functional,application-based arenas. With current deep-learning based protein site specific mutational fitness prediction tools, the direction of “fitness” is usually limited to narrow dimensions, such as protein thermstability. Therefore, with the diversity of selection pressure can be applied individually or in combination, it is likely that distinctin vivoevolution trajectories elucidated will eventually provide protein engineers tremendous prediction powers in a multidimensional protein structure and functional space.
4.2 Directed evolution in higher eukaryotes,including nuclear and organelle genome
At present, effectivein vivoevolutionary system in lower eukaryote is mainly based on segregated,specialized replication system orthogal to nuclear genome. This is mainly because the nuclear genome cannot tolerate high mutagenesis rate asin vivoevolution experiment realistically demands.Therefore, dedicated, local introduction of DNA mutation, in a scenario similar to B cell immunoglobulins gene hypermutation, will be the preferred means to achieve fast, functional diversity.A few CRISPR based local based editing, or nicking induced error prone repair have provided proof-ofprinciple examples of the feasibility of such targeted mutagenesis approach[41]. However, current CRISPR or TALEN based local mutagenesis is very limited in the activity window allowed, due to the underling targeting specificity requirement. On a larger scale similar to V(D)J region of Ig gene, more creative targeting approach must be devised. Unveiling underlying mechanism of activation-induced cytidine deaminase (AID) targeting specificity is going to shed important insights on how to artificially dial transcription-coupled mutagenesis control using ssDNA specific deaminase[42]. On the other hand,dsDNA specific mutagenic enzymes such as DddA can be engineered to confer mutagenic activity only upon targeted onto a specific DNA region, through local protein based phase separation or targeted recruitment by a domain wide scaffold DNA binding protein[43-44]. Similar approaches, once established,can be used to introduce widespread mutation in particular region on the nuclear genome, and even the whole mitochondria and chloroplasts genomes.Regardless of the precise location those local mutagenesis hotspot will be generated, single cellbased phenotyping and live cell tracing capability will be needed for upcoming new era of continuous evolutionin vivo.
AcknowledgementAll figures in this article were created with BioRender.com with the help of XU Hong-En from Zhengzhou University. We are grateful for the assistance and support provided by the Henan Key Laboratory for Pharmacology of Liver Diseases and the Henan Engineering Technology Research Center for Accurate Diagnosis Neuroimmunity for the smooth implementation of this study.
