未折叠蛋白响应的激活机制*
2023-05-16王立堃徐芬芬
王立堃 李 桃 徐芬芬
(中国科学院生物物理研究所生物大分子国家重点实验室,北京 100101)
真核细胞的内质网(endoplasmic reticulum,ER)是分泌蛋白和膜蛋白折叠和翻译后修饰的重要场所。当蛋白质折叠和转运异常时,大量未折叠和错误折叠蛋白会在ER腔累积,这被称为“ER应激”。ER 应激发生时,IRE1、PERK 和ATF6 以不同方式被激活,启动分子伴侣和折叠酶的表达,同时调整蛋白质翻译、转运、降解速率,以恢复ER正常功能。这三条通路被称为“未折叠蛋白响应(unfolded protein response,UPR)”。作为ER应激的感受器,IRE1、PERK 和ATF6 如何感知未折叠蛋白累积这一信号是该领域基本的科学问题。作为ER定位的跨膜蛋白,三者既能对ER腔环境改变也能对ER 膜状态变化作应答。近年来的研究发现,来自胞质和胞外的信号也能激活UPR。三条UPR通路的存在以及各自特有的激活方式使得UPR 信号的调控和功能更加多样化。UPR 与细胞多种生理功能密切相关,也与疾病的发生发展密不可分,对于生理及生理病理条件下UPR 激活机制的深入研究有助于进一步理解UPR 的调控机制和生理意义。这方面还有很多问题亟待解决。
本文对UPR 的激活机制作一介绍,将从未折叠蛋白依赖和非依赖的机制分别阐述。对于UPR和ER 应激的关系,提出对今后需解决问题的看法。
1 UPR
1.1 UPR的发现及其和ER应激的关系
UPR概念是在研究细胞如何应对ER 腔未折叠蛋白累积这一问题中提出的。真核细胞中,分泌蛋白和膜蛋白的折叠和组装在ER里进行,ER腔中高度的氧化环境为蛋白质氧化折叠提供了必要条件[1]。ER也是蛋白质N-连接糖基化的场所,帮助糖蛋白正确折叠的分子伴侣钙网蛋白(calreticulin,CALR)、钙联结蛋白(calnexin,CNX)都是钙结合蛋白,它们的活性受Ca2+调控[2]。可以想见,糖基化受阻,或是ER腔氧化还原水平、钙浓度受干扰会影响ER 腔内蛋白质的折叠。在20 世纪70 年代,人们发现葡萄糖剥夺能特异性激活一系列基因的表达,这些基因的蛋白质产物被称为“葡萄糖调控 蛋 白(glucose-regulated protein,GRPs) ”[3]。20世纪80年代鉴定到ER腔定位的、能够结合免疫球蛋白重链的蛋白质,将其命名为重链结合蛋白(binding immunoglobulin protein,BiP),随即人们认识到BiP 就是GRP 成员中的GRP78[4-5]。利用N-连接糖基化抑制剂衣霉素(tunicamycin,Tm)或葡萄糖类似物2-脱氧葡萄糖(2-deoxyglucose)处理细胞,能上调BiP 表达。不仅如此,BiP 能与错误折叠蛋白结合,而在细胞中表达错误折叠蛋白或过表达分泌蛋白也被证实能诱导GRPs 表达[6-7]。这些实验现象促使人们将葡萄糖剥夺和蛋白质错误折叠联系起来,并提出,细胞具有一种叫“未折叠蛋白响应”即UPR的机制,能对ER腔内错误折叠和未折叠蛋白累积这一信号做出响应,上调GRPs的表达。
错误折叠蛋白在ER 腔累积的现象被称为ER应激。利用Tm 抑制蛋白N-连接糖基化、利用ER膜定位的Ca2+泵SERCA 的抑制剂毒胡萝卜素(thapsigargin,Tg)导致ER腔Ca2+浓度下降,或是利用小分子还原剂如β 巯基乙醇或二硫苏糖醇(dithiothereitol,DTT)破坏蛋白质二硫键,都能造成错误折叠蛋白在ER腔中累积。这也是常用的诱发ER 应激的实验手段[8]。内质网应激下,BiP的上调是在转录水平进行的。酿酒酵母的BiP同源基因KAR2的启动子区具有一个由22个碱基对组成的顺式作用原件,对于Tm 处理下KAR2 转录活性上调至关重要,被称为未折叠蛋白响应原件(unfolded protein response element,UPRE)[9-10]。20 世纪90 年代初期,以Walter 和Mori 为代表的研究者利用UPRE驱动表达的报告系统,在酿酒酵母中进行了一系列遗传筛选工作,鉴定到迄今已知最为保守的UPR 通路:Ire1 及其下游转录因子Hac1[11-14]。1998 年,哺乳动物细胞中Ire1 的同源物ERN1 (也称为肌醇需求酶1α,即inositolrequiring enzyme 1,IRE1α) 和ERN2 (也 称 为IRE1β) 被 发 现[15-16]。2001 年,Hac1的同源物XBP1(X-box-binding protein 1)被鉴定到[17]。有意思的是,在此之前XBP1 已被发现是转录因子,调控B 细胞主要组织相容性复合物II 类分子的表达[18]。后生动物中,除了上述IRE1通路外,还发现另外两条UPR 通路,蛋白激酶RNA 样ER 激酶(PKR-like ER kinase,PERK)和激活转录因子6(activating transcription factor 6,ATF6)[19-21]。至此,目前通常所说的三条UPR通路均被鉴定到。
1.2 UPR信号通路
UPR 信号通路可经由IRE1、PERK 和ATF6 启动。这3 种蛋白质均为ER 定位的一次跨膜蛋白。在哺乳动物中,IRE1α 全身性表达,而IRE1β 在肠道和肺上皮细胞中特异性表达,因此说到IRE1 通路,人们也常只提及IRE1α。一般认为,当ER 腔内未折叠蛋白和错误折叠蛋白累积时,IRE1、PERK 和ATF6 接收该信号并通过不同方式将信号传递到细胞质和细胞核,通过转录水平和翻译水平的调控来帮助蛋白质折叠,恢复ER 稳态。但是,持续激活的UPR 也可能诱导程序性细胞死亡(图1)[22]。
IRE1α 由N 端ER 腔结构域、跨膜区和C 端胞质区组成,其中胞质区包含两个结构域,分别具有蛋白激酶和RNA 内切酶活性。IRE1α 能特异性识别并切割XBP1mRNA,启动XBP1mRNA 剪接,移除一段26 bp 的内含子。这会导致翻译阅读框的移码,其结果是剪接后XBP1(XBP1s,s 指spliced)的翻译推迟终止,产生具有转录因子活性的XBP1s 蛋白[11-12,15]。XBP1s 通过上调与蛋白质转运、折叠、分泌和降解相关基因的表达,以应对ER 应激。IRE1α 还能降解一些mRNA 和微小RNA(microRNA)前体,该过程被称为受调控的IRE1依赖的降解(regulated IRE1-dependent decay,RIDD)。RIDD 导致那些附着于ER 膜胞质侧的RNA(它们往往是ER定位蛋白、分泌蛋白和膜蛋白的mRNA)降解,影响细胞功能,也可能减轻ER蛋白折叠负担[23-25]。 microRNA 前 体 如pre-miR17 的降解则被报道和程序性细胞死亡相关[26-27]。IRE1α也能与肿瘤坏死因子受体相关因子2(tumor necrosis factor receptor associated factor 2,TRAF2) 结 合, 上 调c-Jun N端激酶(c-Jun N-terminal kinase,JNK) 磷酸化并引起细胞凋亡[28],或是促进NF-κB 活化,从而激活炎症因子的产生[29]。
与IRE1α 类似的,PERK 也是I 型跨膜蛋白,其C端胞质区含激酶结构域。PERK的激活也伴随着二聚化的发生。PERK能磷酸化真核翻译起始因子2的α亚基(eukaryotic translation initiation factor 2,subunit 1 alpha,eIF2α),抑制蛋白质翻译,减轻ER 蛋白质折叠的负担。但是,一些5'非翻译区含有上游开放阅读框(upstream open reading frame,UORF)的mRNA,此时主要ORF的翻译水平反而会增强。转录因子ATF4 就是典型的在eIF2α 磷酸化时翻译上调的蛋白质[19-20]。一方面,ATF4 能上调参与氨基酸代谢、抗氧化反应及自噬相关基因的转录;另一方面,ATF4 也能上调GADD34 表达,而后者能负反馈抑制eIF2α 磷酸化。当PERK 持续激活时,ATF4 能上调凋亡相关转录因子CHOP 和死亡受体DR5 的表达,启动细胞凋亡。此外,PERK 还被报道能磷酸化核因子erythroid 2 相关因子 (nuclear factor erythroid 2-related factor 2,NRF2),后者作为转录因子能促进抗氧化反应相关基因的表达。
ATF6 是II 型跨膜蛋白。ER 应激发生时,ATF6 从ER 转移到高尔基体,并被蛋白酶S1P 和S2P 水解,产生的N 端片段(称为ATF6f)从高尔基体上脱落,入核发挥转录因子功能[30-31]。ATF6f和XBP1s均能结合到UPRE区,上调包括BiP在内的UPR 相关基因转录。ATF6f 还能结合到ER 应激元件I/II(ER stress element I/II,ERSE-I/II),上调基因转录[32]。ATF6f还能促进XBP1和CHOP的表达[17,33]。对于ATF6 激活是否也能引起细胞凋亡,目前还不清楚。

Fig. 1 The unfolded protein response and its downstream signals图1 UPR通路及其下游信号
需要指出,上述3 条UPR 通路不是截然分开的。ATF6f 和XBP1s 激活转录的基因有广泛的交集。它们都能通过上调磷脂酰胆碱的生物合成来增大ER 体积,也都能上调ER 膜定位的蛋白质转运体SEC61 复合物的表达[34-36]。不同UPR 通路间也存在相互调控。比如PERK 通路能上调磷酸酶RPAP2 的表达,而RPAP2 可造成IRE1α 的去磷酸化[37]。IRE1α 能降解DR5mRNA,也能降解BiPmRNA,因而对三条UPR 通路下游信号均有负调控[24,38]。此外,XBP1s 和ATF6f 能形成异源二聚体,激活“ER 相关的蛋白质降解(ER-associated protein degradation,ERAD) ”相关基因的表达[39]。这里,ERAD是ER内错误折叠蛋白的降解途径,通过将ER 腔内错误折叠蛋白转运到胞浆,再经由泛素-蛋白酶体系统降解[40]。UPR下游信号非常复杂,影响细胞诸多生理功能,有关UPR 功能的更多研究可参考相关综述[22]。
纵观UPR 的发现历程,可以看到它的研究是和ER 应激分不开的。直到今天,研究者有时还将UPR 和ER 应激这一概念等同起来。本文对当前UPR 激活机制的研究作一介绍,并试图厘清UPR与ER应激的关系,提出该方向有待解决的问题。
2 聚合状态对IRE1活性的影响
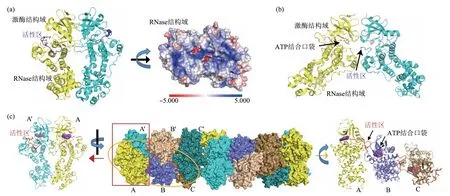
Fig. 2 The crystal structure of the cytosolic part of IRE1图2 IRE1胞质区晶体结构
生化和结构研究揭示了IRE1 二聚化和寡聚化在其活化中的必要性。带有荧光蛋白标签的IRE1在ER应激时能聚集形成点状,说明细胞内IRE1也能发生聚集[41]。IRE1 同源二聚体的形成导致亚基间发生反式自磷酸化[42]。虽然IRE1α 激酶结构域磷酸化对其发挥RNA 酶活性而言并非必需,但激酶结构域处于“激活”构象是必要的[43-44]。这可能导致了IRE1α 的某种构象变化,从而更容易剪切XBP1mRNA。晶体结构显示,酵母Ire1 胞质段以“背靠背”二聚形式存在,C 端有一表面富含碱性残基的区域,推测是XBP1mRNA 结合位点[45-46]。类似的聚合方式在人源IRE1α胞质段晶体结构中也被观测到(图2a)[47]。对人源IRE1α胞质段晶体结构的解析还鉴定到另一种“头碰头”的二聚化状态。在“头碰头”结构中,彼此靠近的亚基激酶结构域中活性中心彼此靠近。IRE1α磷酸化主要发生在一段称为“活性区(activation segment)”的无规卷曲区,虽然活性区由于柔性太大而难以识别,但从附近肽段走向上可以推测活性区从激酶结构域伸出并靠近邻近亚基激酶结构域的ATP 结合口袋(图2b)。这与IRE1α 的反式自磷酸化相吻合。但是,在“头碰头”构象中,亚基的RNase结构域彼此分离,难以解释其如何结合RNA 底物。有可能IRE1α以“头碰头”形式发生反式自磷酸后,再以未知方式转换为“背靠背”模式,发挥RNase 活性[48]。酵母Ire1 胞质段的晶体结构还呈现一种14聚体的螺旋结构,14聚体内部亚基间以“背靠背”模式结合成二体,相邻二体间围绕螺旋轴以右手螺旋方式彼此相错51.4o排列(图2c)。突变破坏“背靠背”二体内部亚基间相互作用位点、抑或破坏相邻二体间结合面都能抑制Ire1 活性[49]。我们注意到,虽然“背靠背”二体内任一亚基的活性区与另一亚基的ATP 结合口袋距离很远,但二体内每个亚基的活性区都与相邻二体中与该亚基相错51.4o的亚基的ATP 结合口袋靠近(最近距离大约10 Å),在14聚体内形成依次衔接模式。考虑到活性区的柔性,推测这种螺旋状聚集体可能是IRE1既有激酶活性又有RNase 活性的形式(图2c)。此外,晶体结构显示IRE1 的ER 腔结构域(lumenal domain,LD)也能形成二体[50-51]。最近利用原位冷冻光电关联显微技术(cryogenic correlated light and electron microscopy combined with electron cryotomography,cryo-CLEM-ET)和免疫电镜技术对细胞内IRE1α聚焦点的观察显示,IRE1α寡聚体位于具有复杂分支的狭窄管状ER,且IRE1α ER腔结构域可能在ER腔内壁附近形成两束相互缠绕的左旋螺旋[52]。总之,IRE1可能有多种聚合状态。寡聚体的形成对于XBP1 剪接是必要的,但RIDD 功能可能主要由二体行使[53]。
3 来自内质网的UPR激活信号
3.1 ER腔内未折叠蛋白累积
这是最为经典的UPR 激活途径。对未折叠蛋白在ER腔中的累积如何被感知并启动UPR,目前主要有直接结合模型和间接识别模型两种解释(图3)。
直接结合模型认为,IRE1 的ER LD 能直接与未折叠蛋白结合并被激活(图3)。酿酒酵母Ire1的LD晶体结构显示,Ire1-LD分子间通过反平行β折叠片层形成二聚体,在二聚体顶部有一长而深的富含疏水残基的凹槽,结构上类似于MHC-I,推测能结合多肽[50]。体外实验证实,Ire1-LD能与富含碱性和疏水残基的多肽结合,凹槽底部Trp426 突变为Ala 则会削弱蛋白质与多肽相互作用[54]。人源IRE1α-LD 晶体结构显示与酵母Ire1-LD 相似的、反平行β折叠片层介导的二聚化,突变破坏二聚化位点能降低IRE1α 活性(图4a)。虽然IRE1α-LD也有一MHC样疏水凹槽,但尺寸太小,不能允许多肽的结合,这对直接结合模型提出了挑战(图4b)[51]。然而,体外实验表明,IRE1α-LD 也能结合多肽,且这种结合能促进IRE1α-LD 寡聚化[55]。IRE1激活的直接结合模型仍需更多生化证据支持。

Fig. 3 The direct association model,competition model,and allosteric model of IRE1α activation under ER stress图3 ER应激下,IRE1α激活的直接结合模型、竞争模型和别构模型
直接结合模型在PERK上也得到了生化和结构生物学实验结果的支持。研究者通过噬菌体展示技术筛选到能和牛源PERK的LD结合的12肽,结晶并解析了PERK-LD 与该多肽复合物晶体结构。结构显示PERK-LD 形成四聚体,其中三个亚基通过一序列保守的凹槽结合12 肽,另一个没有结合(图4c)。通过比较结合和没有结合12 肽的亚基结构,发现12肽诱导结合位点形成β折叠片,在没有结合12 肽的亚基中,这部分由于构象不稳定而在晶体结构中难于被辨认,研究者推测12 肽的结合能稳定该部分构象,而PERK-LD 结合多肽的部位有很大柔性,便于识别和结合具有不同结构的底物蛋白。不仅如此,由于结合12肽而新产生的β折叠片还与位于附近另一个不对称单元的亚基中的β折叠片形成β片层结构,暗示底物蛋白的直接结合能促进PERK 寡聚体形成,而这对于PERK 激酶活性的发挥有促进作用(图4c)[56]。
间接识别模型中,IRE1、PERK、ATF6 在非激活状态下与BiP 结合。当ER 腔出现大量未折叠蛋白时,BiP 从IRE1 和PERK 的LD 区解离,导致IRE1 和PERK 寡聚化(图3)。BiP 的解离是可逆的,这保证了UPR 活化程度的可调控性。在细胞中过表达BiP能抑制IRE1和PERK的活性[57]。BiP结合位点被破坏的IRE1α 突变体在没有ER 应激的情况下也表现出部分活性[58]。以上结果均说明BiP的解离能促进IRE1和PERK活化,但是BiP解离后IRE1 和PERK 的活化是否仍然需要未折叠蛋白的直接结合尚不清楚。另一方面,对酿酒酵母Ire1的研究则得出不同的结果,丧失BiP 的结合位点的Ire1 没有表现出更强的对ER 应激的反应能力,但对乙醇和高温应激更敏感[59]。一种折中的解释是,BiP从Ire1上解离并非Ire1激活的直接原因,但BiP结合Ire1 能促进Ire1 寡聚体的解聚,从而导致Ire1信号衰减[60]。BiP从ATF6上解离也可能是ATF6转移到高尔基体从而被激活的直接诱因,但这只是推测,没有实验证据[61]。

Fig. 4 The crystal structure of the lumenal domain (LD) of IRE1 and PERK图4 IRE1和PERK ER腔结构域(LD)晶体结构
BiP 属于Hsp70 家族,含有核苷酸结合结构域(nucleotide-binding domain,NBD)和底物结合结构域(substrate-binding domain,SBD)。和其他Hsp70 家族成员一样,BiP 与底物蛋白的结合是受NBD 调 控 的:当NBD 和ADP 结合时,SBD 采取“关闭”构象并与底物蛋白形成牢固复合物;在核苷酸交换因子(nulceotide exchange factor,NEF)帮助下,ADP 从NBD 解离并被ATP 取代,此时SBD 成为“打开”状态,底物蛋白释放。一种观点认为,BiP与IRE1结合也是通过SBD,BiP-IRE1结合实际上就是分子伴侣-底物蛋白的结合,这样未折叠蛋白和IRE1 在结合BiP 这一点上是直接竞争关系,这被称为间接识别模型中的竞争模型(competition model)。在该模型中,ERdj4作为BiP的协同分子伴侣(co-chaperone)与IRE1 的LD 结合并招募BiP,后者与IRE1 结合并使IRE1 处于失活状态。ERdj4 也能结合ER 腔未折叠蛋白并将其呈递给BiP,因此,ER 腔中未折叠蛋白会竞争性结合ERdj4 和BiP,削弱它们对IRE1 的抑制作用,导致IRE1活化[62]。需要指出,BiP从IRE1上解离是ATP-NBD 的结合驱动的,而非未折叠蛋白与SBD 结合所致,未折叠蛋白只是与IRE1 竞争性结合那些尚未结合到IRE1 上的BiP。别构模型(allosteric model) 是另一种类型的间接识别模型[63]。在别构模型中,BiP 的NBD 结构域与IRE1结合,其SBD 与未折叠蛋白的结合导致其构象改变,从而从IRE1 上解离(图3)。目前,有关间接识别模型的研究仍主要集中在IRE1 上,上述模型是否适用于PERK和ATF6仍有待验证。
3.2 ER腔定位的UPR活性调控因子
可以看出,间接识别模型中很重要的一点是BiP 对IRE1、PERK 和ATF6 这三种UPR 感受器的抑制。近年来的研究发现了一些ER腔定位的负调控IRE1活性的蛋白质,在IRE1激活程度的精细调控上发挥重要作用。其中,蛋白质二硫键异构酶(protein disulfide isomerase,PDI)家族成员PDIA1和PDIA6 均被报道能抑制IRE1 活性。PDIA1 在被蛋白激酶Fam20C磷酸化后能与IRE1 LD非共价结合,而PDIA6 则与IRE1 LD 的Cys148 形成二硫键[64-66]。Fam20C 是分泌型激酶,其在ER 腔滞留可能是ER应激的信号;PDIA6与IRE1的共价结合则可能是对ER 腔氧化还原状态的某种反映。MANF 是一种内质网应激下表达上调的分泌蛋白,也具有ER 腔定位,可以帮助细胞缓解内质网应激。研究发现MANF 在持续的ER 应激下能结合IRE1,防止其过度激活[67]。ERP47是一种ER腔定位的分子伴侣。与上述分子不同,ERP47能与BiP竞争结合IRE1,且ERP47 的结合能激活IRE1[68]。ER腔定位的UPR感受器调控因子究竟有多少,它们是否以及如何将ER内蛋白折叠状态的信号传递给UPR感受器,有待进一步发现和研究。
3.3 ER膜
不难想到,ER膜状态改变能调控ER膜蛋白的构象和功能。作为ER 定位的跨膜蛋白,IRE1、PERK和ATF6三个UPR感受器均能在ER膜脂饱和度增加的情况下被激活。研究发现,当用棕榈酸(一种长链饱和脂肪酸)处理细胞和抑制酯酰辅酶A 去饱和酶1(stearoyl CoA desaturase 1,SCD1)时,ER 膜脂饱和度增加,此时IRE1α 和PERK 通路被激活,且激活不依赖于两者LD的存在,因而与ER腔内蛋白质错误折叠与否无关[69-70]。对IRE1的进一步研究发现,其跨膜螺旋的N端有一两亲性螺旋,对于感知这种膜状态的变化至关重要。分子动力学模拟显示,跨膜区以一种斜插的方式穿过脂质双分子层,而其N端的两亲性使得这部分附着在ER膜面向ER腔一侧的表面,这种与脂质结合的方式会在其附近压缩膜厚度,使整个系统处于一种高能不稳定状态。跨膜螺旋彼此靠近能缩小这种高能微区,从而释放能量使系统更稳定。脂酰链饱和度的增加及固醇比例的增加使得磷脂双分子层厚度增加且脂质分子排布更有序,此时跨膜螺旋彼此结合能释放更多能量,热力学上讲是更容易发生的。这可以解释为什么ER膜脂饱和度增加能促进IRE1寡聚化(图5a)[71]。另一项研究提出了不同的解释,认为位于跨膜区中部、完全埋藏在脂质双分子层内部的一个色氨酸残基对于IRE1 聚集是重要的,这可以用色氨酸侧链的两亲性解释。将此残基突变为苯丙氨酸残基就足以破坏IRE1 对膜脂饱和度增加的感应(图5b)[72]。对于PERK 如何感知并响应ER膜的改变目前尚未见报道。ATF6的跨膜区能感知ER膜上特定鞘脂二氢鞘氨醇和二氢神经酰胺的存在而被激活,但二氢鞘氨醇和二氢神经酰胺并不激活IRE1和PERK(图5c)[30]。
研究者用“脂双层应激(lipid bilayer stress,LBS)”来命名由于ER 膜脂成分和膜物理性质的变化造成的细胞应激,以区别于ER应激。ER应激下IRE1 能形成光学显微镜下可见的聚集斑点(puncta),但对酿酒酵母Ire1 的研究发现,LBS 不会造成IRE1 聚集斑点形成,所引起的转录组变化也不同于ER应激下转录水平改变[73]。LBS是否使IRE1处于一种和ER应激下不同的激活构象并启动特异的下游信号通路,还需要进一步研究。此外,ER通道Sec61复合物和ER跨膜蛋白EI24也被报道能结合并阻止IRE1 的激活[74-75]。LBS 是否影响上述蛋白质和IRE1的相互作用仍有待回答。

Fig. 5 The activation of IRE1α and ATF6 under lipid bilayer stress(LBS)图5 脂双层应激(LBS)下IRE1α和ATF6的激活
4 来自胞质的UPR激活信号
4.1 UPR感受器结合蛋白
迄今已有不少关于UPR 感受器-胞质蛋白相互作用的报道。这些蛋白质-蛋白质相互作用使得三条UPR 通路的调控具有更好的特异性和灵活性。这方面的研究目前仍主要集中于IRE1。需要指出,胞质蛋白对UPR 感受器的调控是多方面的,这里仅对促进UPR活化的蛋白质做介绍。
Bcl-2 家族是细胞凋亡信号途径中关键的凋亡调节因子。前面讲到,IRE1 的持续或过度激活能造成程序性细胞死亡。Bcl-2家族成员BAX和BAC能与IRE1α 胞质区相互作用并增强IRE1 活性[76],BIM 和PUMA 则在IRE1 下游XBP1 信号的持续激活和RIDD发生中扮演重要角色[77]。非肌肉性肌球蛋白重链IIB(nonmuscle myosin heavy chain IIB,NMHCIIB)能特异结合IRE1α 并帮助IRE1α 聚集体形成[78]。因此,细胞骨架蛋白对UPR 感受器聚合状态的调控可能是UPR 激活的一种新方式。类似的,酪氨酸蛋白激酶ABL1 也能结合IRE1 胞质区并造成IRE1 的寡聚化,这一功能不需要ABL1激酶活性[79]。在上述例子中,NMHCIIB 和ABL1很可能起到支架蛋白的作用,辅助和促进IRE1 寡聚体形成。
UPR 感受器的翻译后修饰是另一种激活UPR的手段。蛋白激酶A(protein kinase A,PKA)是胞质定位的蛋白激酶,在糖脂代谢中发挥重要作用。PKA 能直接磷酸化位于IRE1α 活性区的Ser724,这是IRE1α 上高度保守的磷酸化位点。PKA 是除IRE1 外第一个报道的磷酸化IRE1α 的激酶,其对IRE1α的磷酸化在胰高血糖素信号途径中扮演重要角色[80]。E3 泛素连接酶CHIP(carboxyl terminus of HSC70-interacting protein) 能 催 化IRE1α的泛素化,其中一个泛素化位点Lys545突变为精氨酸残基后会下调IRE1α 的磷酸化。CHIP 催化的IRE1α 泛素化对IRE1α 下游XBP1 信号没有影响,但是能增强IRE1α-TRAF2 互作及其下游JNK磷酸化[81]。这种特异激活IRE1α 激酶结构域、但不影响RNase活性的激活特点是目前已知的、来自ER的UPR激活信号所不具备的。此外,多腺苷二磷酸核糖聚合酶(poly(ADP-ribose)polymerase,PARP)催化多个ADP-核糖分子转移至靶蛋白的翻译后修饰,研究发现PARP16 能聚ADP-核糖基化IRE1α 和PERK,但不会修饰ATF6。在没有ER 应激的情况下,该修饰也能导致IRE1α和PERK的激活[82]。PARP16 是ER 跨膜蛋白,其酶活结构域位于胞质,可见这种修饰发生在胞质侧;但PARP16的C 端位于ER腔部分对于其激活IRE1 和PERK也是必需的。在用Tm 诱导ER 应激发生的前提下,PARP16 能减少IRE1α 和PERK 与BiP 互作,因此PARP16的ER腔段和胞质段对IRE1α和PERK的激活均有作用[82]。
判断一个蛋白质激活UPR是否需要ER腔内未折叠蛋白信号为前提的标准是,该蛋白质是否能激活缺失LD的UPR感受器。遗憾的是,这方面的研究非常少,主要障碍在于过表达的、缺失LD 的IRE1 和PERK 能自发激活。如果通过基因编辑技术直接改造内源基因,对于回答这一问题将有很大帮助。
4.2 Ca2+离子
ER是细胞内的“钙库”,其腔内Ca2+浓度上千倍于胞浆中Ca2+浓度,ER膜定位的钙泵SERCA和钙通道蛋白IP3R、RyR等能调控ER内外Ca2+浓度,影响ER稳态和细胞生命活动。破坏ER的Ca2+稳态是常用的ER应激诱导方式之一,但是对于这究竟如何激活UPR,目前并不清楚。最直接的可能是ER 腔依赖的蛋白质折叠受阻,导致未折叠蛋白和错误折叠蛋白的大量累积。值得注意的是,近期的研究发现,胞质Ca2+浓度上升也可能是PERK活化的直接因素。SERCA 活性受抑制时,胞质Ca2+浓度的升高能迅速上调PERK磷酸化水平,甚至对于缺失LD 的PERK 也是如此[83]。进一步研究揭示,Ca2+能直接结合并稳定PERK 胞质段,但Ca2+对PERK磷酸化的促进仍然需要Mg2+,后者是激酶辅因子[84]。Ca2+究竟以何种方式结合PERK,又是如何调控PERK构象变化,还需要进一步研究。
5 来自胞外的UPR激活信号
5.1 细胞膜受体介导的UPR激活
生理水平UPR 的激活不局限于单个细胞,来自胞外的信号也能激活UPR。Toll 样受体(Tolllike receptor,TLR)在感知病原入侵和激活免疫应答中发挥着重要作用。巨噬细胞的TLR 通路能特异性激活IRE1α 及其下游XBP1mRNA 剪接,进而促进细胞因子产生[85-86]。TLR4 和TLR2 通过诱导NADPH 氧化酶2(NOX2)的表达来激活IRE1α,但具体分子机制未知[85]。另一项研究显示,肿瘤坏死因子受体关联因子6(tumor necrosis factor receptor-associated factor 6,TRAF6)在TLR 介导的IRE1α活化中起重要作用,TRAF6造成IRE1α泛素化,而这抑制了IRE1α与催化其去磷酸化的磷酸酶PP2A 的相互作用,导致IRE1α 活性上升[86]。XBP1是在研究B细胞抗原呈递时发现的[18]。考虑到IRE1α-XBP1通路在免疫反应中的重要性,其处于TLR 信号下游并不奇怪。TLR 信号激活IRE1α-XBP1还能上调前列腺素2 (prostaglandin H2,PGH2)的生物合成,在小鼠的痛觉反应中发挥重要功能[87]。
血管内皮生长因子(vascular endothelial growth factor,VEGF)可在体内诱导血管新生。VEGF 能快速激活人脐静脉内皮细胞(HUVEC)中IRE1α、PERK 和ATF6 三条UPR 通路,体内实验表明UPR 的激活对于血管新生至关重要。机制研究发现,VEGF 是通过激活PLCγ、进而激活mTORC1 来促进UPR 活化的。虽然没有明确的证据,但推测VEGF 受体参与了VEGF-PLCγmTORC1-UPR信号传递[88]。
脑源性神经营养因子 (brain-derived neurotrophic factor,BDNF)是神经营养因子家族成员之一,在神经元的分化、成长与重塑中起重要作用。BDNF 能在神经元中特异性激活IRE1α-XBP1信号通路[89]。和IRE1α在对胰高血糖素信号应答中一样,BDNF也是通过促进PKA对IRE1α的磷酸化实现的[90]。推测BDNF 的受体原肌球蛋白相关激酶B(tropomyosin-related kinase B,TrkB)在BDNF激活IRE1α的过程中起作用。
5.2 细胞非自主的UPR
细胞非自主的UPR是近年来提出的一个概念,指细胞将ER应激信号以某种方式传递到其他细胞并在信号接收细胞中激活的UPR。这一现象已在不同类型细胞、组织和器官中被观察到,包括秀丽线虫的神经细胞-肠道细胞、小鼠下丘脑POMC 神经元-肝脏细胞、肿瘤细胞-肿瘤细胞、肿瘤细胞-免疫细胞、心肌细胞-巨噬细胞、肝细胞-肝细胞、肌细胞-肌细胞等[91-102]。其中一些研究鉴定到供体细胞的IRE1α-XBP1信号是产生细胞非自主UPR的必要条件,但这是否适用于所有情况还有待检验[93-94,98]。对于跨细胞信号传递方式,有研究认为分泌因子以小细胞外囊泡(small extracellular vesicle,sEV)形式传递,比如在肌细胞和肌细胞间的传递[102]。也有报道称肝细胞-肝细胞之间是通过细胞间连接传递信号的[99]。但更多的研究中并未对该问题作出清晰的回答。
哪些分子的跨细胞传递导致细胞非自主UPR的发生?在秀丽线虫中,神经细胞分泌的酪胺能激活肠道细胞IRE1-XBP1 通路[98]。棕榈酸刺激导致ER 应激发生的肌细胞则通过传递神经酰胺到下游肌细胞,激活下游细胞IRE1α-XBP1 通路[102]。在肝细胞癌中,发生ER 应激的癌细胞通过上调高尔基蛋白73(Golgi protein 73,GP73)的分泌,激活肿瘤浸润的巨噬细胞UPR[97]。
从以上例子可以看出,细胞非自主UPR 可能没有统一的激活机制,而是存在细胞和组织特异性。在不同的ER 应激条件下,细胞也可能以不同的方式启动UPR 信号的跨细胞传递。该领域还有很多问题有待深入研究。
5.3 其他环境因素
UPR 与肿瘤的发生发展密切相关,大量研究报道肿瘤细胞中存在UPR 的激活。肿瘤细胞自身的基因突变是UPR 活化的原因之一,肿瘤细胞所处环境的特殊性也可能在UPR激活中起重要作用。缺氧是肿瘤微环境的特征之一,细胞培养中低氧应激能导致UPR,但这往往需要很低的氧分压(1%或更低),在接近生理病理条件(1%~5% O2)条件下UPR 只有微弱的激活。缺氧干扰了蛋白质的氧化折叠,造成ER 应激;氧气对于脂质去饱和化也是必需的,脂质饱和度增加会影响ER体积的扩张,不利于ER应激的缓解,此外也会造成LBS[71,103]。缺氧导致IRE1-XBP1 信号通路激活,研究发现XBP 可以 与缺氧诱导因子1α (hypoxia-inducing factor 1α,HIF1α)结合,共同促进HIF1α 靶基因的转录[104]。缺氧会下调蛋白质翻译,这对于肿瘤细胞而言是一种保护措施,PERK的缺失不利于翻译水平的下降[105]。缺氧也会造成细胞活性氧水平上升,干扰蛋白质糖基化[106]。PERK 可以诱导谷胱甘肽合成,从而降低缺氧应激下细胞活性氧水平[107]。
酸性环境是另一种造成UPR 的因素。肿瘤细胞糖酵解产生乳酸造成pH 值下降,乳酸和低氧共同刺激下肿瘤细胞XBP1、CHOP 和ATF3 表达上升[108]。在内皮细胞中,微酸性环境(pH 6.4)能导致eIF2α 和IRE1α 磷酸化,上调ATF3、ATF6f 和XBP1mRNA 剪接水平[109]。质子感应G 蛋白偶联受体GPR4 和GPR68 在酸性激活UPR 中起作用[109-110]。
葡萄糖缺乏和过多也被报道能激活UPR。葡萄糖缺乏影响蛋白质糖基化,可以造成ER 应激[111]。葡萄糖缺乏影响胰岛β 细胞的SERCA 活性,干扰ER腔Ca2+稳态,促进PERK和eIF2α磷酸化[112]。高糖刺激也能激活β 细胞IRE1α 通路,但具体机制尚不清楚[88]。代谢相关疾病如糖尿病、肥胖等,往往伴随着UPR 信号上调,这是否能归结于高糖高脂刺激还有待研究。
UPR 与很多疾病密切相关,在癌症、代谢性疾病、神经退行性疾病中均有UPR 通路激活的报道[113]。以上举例说明肿瘤和代谢疾病中环境因素导致UPR 激活。需要指出,生理和生理病理条件下UPR 的研究更多关注其生理意义,忽视了对UPR 激活机制的细致探究。复杂环境下UPR 的激活很可能是多种因素综合作用的结果。
6 讨论和展望
迄今为止,IRE1 是了解最多的UPR 感受器,也是已知的唯一能切割XBP1 mRNA、启动XBP1 mRNA 剪接的蛋白质。已知的PERK 和ATF6 下游信号则并非仅位于这两个UPR 感受器下游。尤其需要注意的是,PERK-eIF2α 通路属于整合应激响应(integrated stress response,ISR)的一支,ISR还包括另外3 条通路:PKR、GCN2 和HRI。在氨基酸缺乏、线粒体应激、氧化应激等条件下,ISR通路的激活均可引起eIF2α 磷酸化,继而上调ATF4、CHOP的表达(图6)[114]。一些研究将上述ISR 下游信号归结于PERK 激活,但并没有验证PERK磷酸化水平是否上升,这有可能得出错误的结论。
UPR 和疾病密切相关,但对生理病理条件下UPR 激活机制的研究很困难,这一方面是因为这些条件下UPR 激活很可能是多种因素综合作用所致,另一方面也因为研究者更关注UPR 激活的生理意义。事实上,对UPR 生理功能的认识很多来自UPR 感受器基因敲除实验,缺乏严格的实验说明,在野生型细胞或个体中相同的生理条件下UPR感受器确实被激活。
UPR 与未折叠蛋白的关系是另一个值得关注的问题。虽然最初的UPR概念是指细胞对ER内未折叠蛋白累积这一信号的应答,但越来越多的研究发现,UPR 也能以不依赖于未折叠蛋白的方式被激活。厘清UPR激活与未折叠蛋白累积间的关系,对于深入理解UPR 激活机制至关重要。然而,目前缺乏实用的检测ER 腔内未折叠蛋白和错误折叠蛋白的工具。硫黄素T(thioflavin T,ThT)染料与蛋白质聚集体结合后荧光信号增强,被用于检测蛋白质聚集体,但该染料更适用于检测淀粉样沉淀[115-117]。有研究表明,ThT能用于检测ER内未折叠蛋白聚集,其实用性尚需更多的实验检验[117]。近期报道一种ER 腔定位的Halo 蛋白突变体(AgHalo(ER)),这种突变体容易发生错误折叠和聚集,同时能被小分子荧光探针标记,因而可用于指示ER蛋白质稳态[64,118]。但AgHalo(ER)只对热应激有反应,且长时间表达会自发聚集,限制了其应用[118]。开发更好的检测ER 腔内蛋白质稳态的工具对于推进UPR机制研究很有必要。

Fig. 6 The four signal pathways of the integrated stress response图6 整合应激响应的四条信号通路
