荧光核酸适配体:核酸酶学分析新机遇*
2023-05-16高子珩薛婷婷陈显军
高子珩 邹 譞 周 宜 薛婷婷 陈显军** 杨 弋**
(1)华东理工大学,生物反应器工程国家重点实验室,光遗传学与合成生物学交叉学科研究中心,上海 200237;2)华东理工大学药学院,上海市细胞代谢光遗传学技术前沿科学研究基地,上海 200237)
核酸的代谢过程遵循着一系列的规律,包括自我复制、修饰、转录、剪接、转运、翻译和降解。某些病毒中的逆转录和高等动物特有的端粒复制也是核酸代谢过程的一部分。核酸代谢不仅是生物体正常生长发育、遗传变异和细胞分化的重要因素,而且还与癌症、辐射损伤、病毒感染、遗传病和代谢病等疾病的发生和发展有着密切联系[1-4]。因此,与核酸代谢相关的酶是维持生物代谢和细胞内稳态的重要角色。
DNA 解旋酶BLM、WRN 和RECQ4 的异常可能会导致许多罕见的疾病,包括布卢姆综合征(Bloom syndrome,又称面部红斑侏儒综合征)、沃纳综合征(Werner syndrome,又称成人早老综合征)和罗斯蒙-汤姆森综合征(Rothmund-Thomson syndrome),甚至肿瘤[5-6]。拓扑异构酶的异常会对大脑发育过程中的遗传机制产生深远影响,可能导致自闭症谱系障碍(autistic spectrum disorder,ASD)的发生[7];核糖核酸酶RNase H2 则可以激活先天免疫模式,识别受体,引发炎症反应,从而影响大脑的发育过程;爱卡迪-古蒂综合征(Aicardi-Goutieres sydrome,AGS)[8-9]是一种儿童炎症性疾病,它会导致端粒缩短,这种缩短会提高患心血管疾病、肝硬化、高血压症状、动脉粥样硬化、骨髓衰竭、高血糖和肿瘤等疾病的风险[10-11]。
因此,酶活性水平的改变已被用作许多病理研究的指标和主要的研究靶点。某些常见的靶标酶,如DNA 聚合酶、甲基转移酶、拓扑异构酶和解旋酶,对于核酸的代谢过程具有重要的调节作用,可以促进细胞生长与个体发育。同时,核酸代谢相关酶是DNA 测序、分子克隆、定点诱变、表观修饰和聚合酶链反应等生物技术和生物工程领域不可或缺的工具。本文将参与核酸体内代谢过程的相关酶进行了整理与分类(表1)。
1 传统核酸酶学分析方法
建立核酸代谢酶的活性检测方法对于疾病的诊断、临床治疗和病理研究具有重要意义。酶的标准活性检测方法一般是通过测定合成或降解核酸底物的量来进行测定的。传统的核酸酶学分析方法包括比色法[12]、高效液相色谱法[13]、电化学法[14]、凝胶电泳法[15]、放射性标记法[16]和酶联免疫吸附法[17]等。本文对传统的核酸代谢酶检测方法的优缺点进行了整理比较(表2)。

Table 2 Comparison of traditional nucleic acid metabolism enzyme detection methods[18-41]表2 传统的核酸代谢酶检测方法比较[18-41]
2 基于荧光法的核酸酶学分析探针
1962 年2 月,日本籍海洋生物学家下村修[18]在水母体内发现了一种特殊的蛋白质,它在紫外光照射下会产生一种耀眼的绿色荧光,这种蛋白质被命名为绿色荧光蛋白(GFP)。Tsien 教授[19]进一步改良了它,并开发出了一系列具有不同颜色的荧光蛋白,以满足不同的应用需求。在此基础上,越来越多的荧光基团陆续被报道,这些荧光基团被广泛用作酶活性检测中的报告分子。基于荧光的分析方法具备价格便宜、使用简便、敏感度高的优势,而且可以通过多种荧光基团的组合实现正交检测。经过多年的发展,基于荧光法的酶活性分析检测成为实现酶活性快速实时、准确、低成本检测的重要技术。
到目前为止,基于荧光的核酸酶学分析探针可以大致分为3 类:非特异性结合核酸的荧光探针,基于荧光共振能量转移(fluorescence resonance energy transfer,FRET)原理的特异性探针,以及遗传编码的荧光DNA 适配体/荧光RNA 适配体的特异性探针。按照时间的发展顺序,分别称其为第一代、第二代和第三代。
2.1 第一代荧光探针:非特异性结合核酸的荧光探针
核酸染料是一类在结合核酸之后荧光显著增强的小分子化合物,通常具有苯环或共轭双键等离域π键体系提供用于接收激发光的核外电子,同时还要有与DNA特异性相互作用的基团。如图1所示,这种相互作用力主要可分为插入DNA 的碱基之间以及与DNA 双螺旋的小沟相互作用,依赖于前者的代表性核酸染色染料为溴化乙锭(ethidium bromide,EB),依赖于后者的代表性核酸染色染料为Hoechst系列染料。
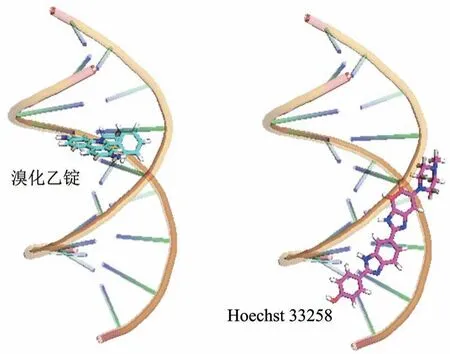
Fig. 1 Schematic diagram of non-specific nucleic acid dyes图1 非特异性核酸染料与双链DNA结合示意图
EB与核酸结合时荧光增强[20]并可用于电泳凝胶中的DNA检测[21],是分子生物学中应用最广泛的技术之一。但是,使用EB 染色的背景荧光高,而且易导致DNA 突变,具有核酸毒性,不利于实验人员的健康。碘化丙锭(propidium iodide,PI)是一种溴化乙锭的衍生物,它能够吸收紫外光,产生明亮的荧光,通过PI与吖啶橙(acridine orange,AO)联用可以鉴别死/活细胞,活细菌会产生明亮的绿色荧光,而死细菌则会产生鲜艳的红色荧光[22]。7-氨基-放线菌素D(7-amino-actinomycin D,7-AAD)是1974 年合成的染色剂,它可以用来检测染色体的结构和功能。7-AAD 对于染色质的结合不受染色质凝聚程度的影响[23],可以区分活细胞、凋亡细胞和死亡细胞[24],其发射波谱较PI窄,对其他检测通道的干扰更小,在多色荧光分析中是PI 的最佳替代品。此外,荧光素二乙酸酯和PI 也可以用于细胞染色,以更准确地鉴定死亡细胞。SYBR Green I 由Molecule Probes 公司开发,并于1997 年被首次应用和报道用于细胞计数[25]。SYBR Green I 已被广泛应用于多种DNA 检测和分析技术,其中包括流式细胞术[25]、实时荧光定量聚合酶链式反应(qPCR) 以及其他多种应用中[26]。SYBR Green I与双链DNA相互作用后可显著增加亮度[27]。然而溶液的酸碱度会明显影响SYBR系列的染色效果,如果溶液pH高于8.3或低于7.5,它的检测灵敏度将会显著下降[28]。Pico Green 染料和SYBR Green I 具有相似的结构和光谱,由Molecule Probes 公司开发,荧光关于双链DNA浓度的线性关系范围从双链DNA浓度25 ng/L至1 mg/L。即使在盐、蛋白质、聚乙二醇、尿素、氯仿、酒精和琼脂糖等存在的情况下,该染料测定的结果也能保持稳定[29]。Gel Red、Gel Green 是EB的结构类似物,通过插层作用结合DNA[30],但是由于是两分子桥联的产物,因此该染料分子质量大,难以透过细胞膜,对DNA和RNA的迁移影响小。Super Gel Blue(US EVERBRIGHT INC)无致突变性,可用蓝光成像避免紫外伤害,热稳定且无需避光储存,可用于琼脂糖及聚丙烯酰胺凝胶电泳中DNA或RNA染色。
Hoechst 33258 和Hoechst 33342 长时 间 暴露 在紫外光下不仅会导致光漂白,还会导致其在蓝光的激发下发射出以绿色为主同时带有黄色和橙色的荧光,可能会对同时使用Hoechst染料与GFP 的实验带来干扰和影响[31]。SiR-Hoechst 是一种使用远红外光激发的核酸染料,由基于硅罗丹明的荧光团(SiR)和能与DNA 小沟结合的基团双苯甲酰亚胺组合而成。结合双链DNA后在670 nm处的荧光增强了50 倍,并且与双链RNA 集合几乎没有荧光,除此之外,应用于活细胞染色时,使用SiRHoechst 对细胞的影响相比于SYTO 61、Vibrant Dye cycle Ruby 和DRAQ5 等DNA染料更小[32]。DAPI是一种DNA序列特异性的染料,其通过附着在富含腺嘌呤-胸腺嘧啶的DNA序列的小沟中形成荧光复合物,此外还可以与仅含有鸟嘌呤-胞嘧啶碱基组成的DNA结合形成无荧光复合物[33]。DIPI是DAPI的结构类似物,但是DIPI对于紫外光照射有更好的抗性,在30 min 内几乎不会褪色[34]。Hoechst系列染料可以方便地与其他常用的红色/绿色荧光基团联用,对活细胞进行正交染色。AO早在1967 年之前就被应用于核酸染色[35],其可以通过染色后的颜色区分DNA和RNA。该染料对DNA染色呈绿色而对RNA 染色则呈现红色荧光[36]。AO能透过细胞膜,可以对活细胞及死细胞内的核酸进行染色[37]。普卡霉素(mithramycin)可以与DNA中富含鸟嘌呤-胞嘧啶的区域结合,并且这种结合主要是这两种化合物从溶液中疏水转移到DNA 的小沟中,吸收470 nm 波长的青光并发射554 nm 的黄光,褶皱霉素和色霉素与普卡霉素拥有相同的核心三环结构,而连有不同的寡糖链,因此其一样可以与DNA的小沟结合,而吸收/发射光谱略有不同[38]。
还有一类专门用作溶液中DNA 定量的核酸染料 如 Quant Fluor (Promega)、 Accu Clear(Biotium)、Qubit(Life Technologies)等核酸定量试剂盒。这些商业试剂盒除了实现DNA及RNA的高灵敏定量进而分析核酸代谢的相关酶的酶活性[39]之外,还在辅助RNA 样品提取[40]、生物气溶胶分析[41]、药物筛选[39]、高通量测序文库构建[42]、类器官培养[43]等方面提供了基础平台。To-Pro-3 是一种噻唑橙的衍生物,是一种可以由642 nm 激光激发的红色荧光团[44]。它无法穿过细胞膜,可以在流式细胞术中染色死亡细胞[45]。To-Pro-3及其二聚体TOTO-3与双链和单链DNA间存在多种相互作用力,有很强的结合能力[46]。DRAQ5 是一种红外荧光双烷基氨基蒽醌染料,对DNA具有高亲和力且可以进入活细胞[47]。它的最大吸收峰在646 nm,最大发射峰在681 nm。但是DRAQ5浓度高至1 μmol/L时会引起核结构的变化,造成染色质的异常聚集问题[48]。SYTO 是一组跨越广域光谱范围的染料,涵盖广泛的可见光激发和发射光谱。所有SYTO染料在无细胞系统中均显示出低本底荧光,与DNA或RNA结合后荧光显著增加[49],量子产率增加了近40 倍[50]。Vibrant Dye cycle Ruby 是由Molecule Probe 公司开发的Vibrant Dye cycle Ruby 染料家族的新型低细胞毒性红色荧光染料,主要用于流式细胞术中的细胞染色[51-52]。
综上,我们对常见核酸染料的优缺点进行了比较(表3)。相比放射性同位素标记,使用核酸染料不仅同样可以在染色后的电泳凝胶上直观地展示核酸的浓度和分布,而且无需繁琐的预处理和针对放射性元素的防护。当核酸染料用于液体样品的检测时,使用酶标仪即可代替昂贵的液体闪烁计数仪。正因如此,在如今的研究中核酸染料已经几乎完全取代了放射性同位素标记法成为主流。但是核酸染料也有着自身的局限性,由于核酸染料是非序列依赖性的,因此在对核酸进行标记时无法区分序列不同的核酸,也无法通过染色区分某一特定的引物的延伸产物,同时,使用核酸染料进行实时荧光定量PCR 时也不得不考虑假阳性问题等。为了摆脱以上种种困境,以TaqMan探针为代表的使用荧光基团标记的寡核苷酸探针被开发出来,这些经过设计的序列以FRET原理为基础,成为了第二类基于荧光的核酸代谢相关酶研究方法。

Table 3 Comparison of advantages and disadvantages of common nucleic acid dyes表3 常见核酸染料优缺点比较
2.2 第二代荧光探针:基于荧光共振能量转移(FRET)原理的特异性探针
第二代荧光探针基于FRET的原理,它能够将分子间的能量以非辐射的方式转移到另一个荧光团,这种转移过程受到分子间距离的限制。这种技术可以有效地检测出分子间的相互作用,从而提高检测的灵敏度和准确性。当供体和受体之间相距在1~10 nm区域内时,由于供体发出的荧光能量被受体吸取,导致供体荧光强度显著降低,而受体荧光强度则会显著增强[53]。
第二代荧光探针可分为化学水解型荧光探针、核酸杂交型荧光探针和分子互作型荧光探针[54]。化学水解型荧光探针中的猝灭基团吸收来自报告基团的荧光信号。在扩增过程中,寡核苷酸与互补目标序列杂交后利用核酸酶的酶切活性,探针中的磷酸二酯键被水解,报告基团和猝灭基团分离,从而释放出荧光信号,如TaqMan[55]、QZyme[56-57]等探针。核酸杂交型荧光探针通过与标记了报告基团的探针与目标DNA 结合从而实现报告基团与猝灭基团的分离,比如双杂交探针[58]、分子信标(MB)[59]、MGB Eclipse 探针[60]、蝎型探针[61]、Ampli-fluor 探针[62]和LUX 引物[63],它们会在延伸过程中无损释放,报告基因和猝灭剂之间建立物理距离,释放报告基因的荧光信号。分子互作型荧光探针的报告基团在连接到RNA 上后不会立即被激活,只有当RNA 与相关的蛋白质结合之后,才会发出强烈的荧光,如PIFE探针[64]。
2.2.1化学水解型探针
化学水解型探针通常用于实时定量PCR。当扩增未开始时,猝灭基团可以有效地吸收报告基团的荧光信号,在扩增步骤中,基于热稳定型Taq聚合酶5'-3'的核酸外切酶活性[55],短寡核苷酸与互补目标序列杂交后被水解,报告基团和猝灭基团分开,进而释放出荧光信号,实现实时检测核酸扩增的目的(图2c)。TaqMan 探针可以在指数扩增阶段准确地定量产物[65-66]。得益于其高特异性,除实时荧光qPCR,TaqMan探针还可用于量化基因表达,遗传多样性等检测,也能够用于各种核酸酶的活性及抑制检测,如靶向核酸切割[67]、糖基化[68]、甲基化[69-70]和磷酸化[71]的酶。
由BD Biosciences 公司2003 年研发的QZyme Assay的原理与其他定量PCR系统的原理相似,但涉及不同的荧光信号生成机制[56-57]。DNAzyme 是一种具有独特催化活力的寡核苷酸,它具有两种底物识别结构域,能够在特殊的磷酸二酯键处有效地切割核酸底物(图2a),QZyme 系统包括了DNAzyme 以及与其非活性链相连的5'端特异性引物和3'端特异性引物各一条,还有一种DNAzyme特异性的荧光底物,这是一种一端标记有报告基团,另一端标记有猝灭基团的寡核苷酸片段。在扩增过程中,产生包含活性的正向DNAzyme 拷贝的扩增子,从而裂解底物,在空间上分离荧光团和猝灭剂,因此扩增子的积累伴随着荧光的增加。
2.2.2核酸杂交型探针
双杂交探针,即相邻杂交探针,由Heller 和Morrison[58]于1985 年首次提出。该测定使用两个与相邻DNA 序列杂交的寡核苷酸探针。两个探针末端分别用发色团标记,其中一个探针有3'供体,另一个有1 个5'受体。FRET 在双杂交后形成,通过供体的猝灭和受体荧光的敏化发出荧光信号。两个荧光基团的激发光光谱有一定程度的重叠,且两条探针需要与靶核酸的杂交位置应相互邻近(通常为1~5 个碱基)。两个探针需要被精确地杂交到特定的靶顺序上才能够探测到荧光,因此该方法具有极强的特异性(图2b)。
1996 年,Tyagi 和Kramer[59]首次提出了分子信标。分子信标是由15~30 个碱基组成的DNA 序列,两侧分别有2个短的互补茎序列,当整个序列在缺少目标序列时,将自发形成茎环结构。茎环结构的形成可以令猝灭剂和荧光团之间更加紧密地结合,有效地抑制荧光信号。当目标DNA或RNA分子存在时,它们之间会发生杂交,杂交越强,荧光团和猝灭剂之间的空间分离更加明显,从而提高荧光信号的强度(图2d)[72]。相比于TaqMan 探针,分子信标在扩增过程中具有更高的稳定性,不会出现降解现象,而且在每个循环中,它都能够与靶标序列结合,从而产生可测量的荧光信号。这种基于分子信标设计的荧光响应目标酶的检测探针已应用于监测各种核酸酶的催化过程,如切割[73-75]、连接、糖基化[75]、甲基化[76-77]和磷酸化[78-79]。
Afonina 等[60]于2002 年 报 道了MGB Eclipse probe,其主要特征是在5'端DNA 小沟结合基团(minor groove binder,MGB)配体以及猝灭剂,在 3'端具有荧光团。单链MGB Eclipse 探针的荧光在未杂交时被末端染料和猝灭基团的相互作用而有效猝灭。在与互补目标杂交后,MGB 分子嵌入双链并使探针高度稳定,这使更短、更特异的探针序列成为可能。5'-MGB-quencher 基团还可以防止探针在PCR 过程中被Taq DNA 聚合酶水解。如图2e所示,归因于MGB和碱基杂交赋予的出色特异性,MGB Eclipse 探针可广泛应用于实时荧光qPCR。MGB Eclipse probe 系统显著地增强特异性,尤其是对于某些特定类型的单核苷酸多态性识别,如富含鸟嘌呤-胞嘧啶的错配、或富含腺嘌呤-胸腺嘧啶的目标核酸中鸟嘌呤-胸腺嘧啶替换,以及任何情况下的鸟嘌呤-胸腺嘧啶错配。
David Whitcombe 团队[61]于1999 年首次报道蝎型探针(scorpions probes)。蝎型探针包括一段目标片段特异性序列、一对自互补的回文序列和一对荧光团/猝灭剂,此外增加了一段与相应靶核酸结合的PCR 引物上,并增加PCR 终止子以防止5'方向茎环序列的复制。从引物进行一轮PCR 延伸后,新合成的目标区域与探针上的特异性序列互补(图2g)。经过第二轮变性和退火处理后,蝎型探针与目标杂交,荧光团与猝灭剂分离,从而释放出强烈的荧光信号,这一特性比TaqMan 探针和分子信标更加迅速、更有效。
Nazarenko 等[62]于2002年首次提出LUX(Light Upon eXtension)探针。LUX 探针基于寡核苷酸的一级和二级结构对共轭荧光团发射特性的影响,只有探针在与PCR 产物杂交形成双链时,共轭荧光团才会增加荧光强度。LUX 探针设计由以下元素构成:以C或G作为引物的3'端核苷酸,荧光团连接至从3'端起的第2 个或第3 个碱基(T),在标记核苷酸两侧的3 个核苷酸内的一个或多个G,以及茎环结构的发夹引物。LUX探针的5'端在低于其熔点的温度下形成平末端发夹。LUX 探针的设计十分简单,可以通过将引物的上述特征和其他如长度和熔解温度(Tm)的特征输入专门的软件,来产生位于整个目标序列中的大量引物对[64]。
2.2.3分子相互作用型探针
对于RNA 及相关靶向酶的检测,蛋白质诱导荧光增强 (protein-induced fluorescence enhancement,PIFE) 能够补充FRET 对蛋白质-RNA 互相作用的检测。PIFE 能够记录蛋白质与RNA 结合后的运动和活性,且PIFE 可以通过全内反射荧光显微镜观察和监测,并只需要一种染料就可以标记一条RNA链(图2f)。PIFE的好处在于它可以节省设计染料结合位点的时间,它不会使染料影响蛋白质的结合亲和力和解离。通常,染料位于RNA 的5'端或3'端,染料的选择也类似于FRET,需要考虑量子产率、荧光强度、寿命、动态各向异性和光稳定性。与FRET相同,PIFE也具有距离依赖性,并且在荧光团和蛋白质附近的0~3 nm 范围内表现更好,而FRET 在3~10 nm 处表现出最高的灵敏度。这表明FRET 和PIFE 在短距离测量RNA-蛋白质复合物动力学方面能够彼此补充[80]。总之,PIFE 可广泛应用于研究RNA 与不同酶如聚合酶、解旋酶或核酶和核糖开关的相互作用,以实现RNA识别和酶活性的检测。

Fig. 2 Schematic of specific probe based on FRET principle图2 基于荧光共振能量转移原理的特异性探针原理图
2.3 第三代荧光探针:基于遗传编码的荧光RNA适配体和荧光DNA适配体
为了开发低成本、高通量、快速的检测核酸代谢相关酶的方法,研究者们进行了一些新的探索。荧光蛋白可在基因水平上进行遗传编码,并在细胞自身生化反应机制下产生荧光。将荧光蛋白与目标蛋白融合可以帮助研究人员“照亮”目标蛋白,揭示目标蛋白在活细胞与活体中的各种行为,为研究细胞内蛋白质在生命活动中的作用提供技术支持,即应用荧光蛋白检测蛋白质的代谢过程,以及蛋白质代谢相关酶的活性。受到荧光蛋白的启发,基于荧光核酸适配体的检测方法被开发出来以应用于对核酸代谢酶活性的研究。
何谓“荧光核酸适配体”?荧光核酸适配体指的是通过合成生物学技术得到的可以与荧光团染料特异性结合并可以激活染料荧光的一类核酸适配体[81]。荧光核酸适配体在自然界中并不存在,必须通过人工合成与筛选的方式得到[82-83]。筛选荧光核酸适配体所使用的技术为指数富集的配体系统进化技术(SELEX)。研究人员构建出长度20~100 nt(丰富度可以达到1014~1015)的单链寡核苷酸文库,随后需要依次经过与靶标(染料小分子)的孵育、洗杂、洗脱、富集、次级文库的制备等多个步骤作为一轮筛选,一般经过6~15 轮的筛选后即可得到与靶标特异性结合的序列。核酸具有制备简便、结构修饰容易、选择性高、亲和力强等多种优势。荧光核酸适配体按照其化学本质可以分为荧光RNA适配体和荧光DNA适配体。
2.3.1荧光RNA适配体
荧光RNA 适配体孔雀石绿适配体(MG aptamer)由Grate 和Wilson[84]于1999 年报道。染料MG结合与该适配体结合后,其最大吸收波长产生了14 nm的红移。2003年,Babendure等[81]发现MG Aptamer可以结合孔雀石绿(MG)及其衍生物并显著增强它们的荧光,荧光激活倍数有的可达2 300 倍以上,据此提出了荧光核酸适配体这一概念。本文对已报道的荧光RNA 适配体及其发色团按照复合物的发射波长进行了总结(图3)。研究者们基于传统染料如Hoechst、噻唑橙、二甲基吲哚红和磺基罗丹明B 等开发了诸如Hoechst 1c[85]、Mango[86]、Peach[87]、DIR2s-apt[88]、o-Coral[89]和SRB-2[90]等RNA荧光适配体。
2008年,Sando等[85]合成了一种Hoechst衍生物Hoechst 1,随后筛选获得了该染料的RNA 适配体I-mini3-4,其荧光激活倍数近30 倍,平衡解离常 数(KD) 为35 nmol/L。Dolgosheina 等[86]在2014 年筛选得到了可以特异性识别并结合TO1 的RNA适配体Mango。Mango以很高的亲和力(KD=3 nmol/L)结合TO1-biotin 并发出黄色荧光,结合TO1 的衍生物TO3-Biotin 产生红色荧光。2008 年,Constantin 等[91]筛选获得了称为DIR-Apt1 的RNA适配体,它与DIR 结合的KD为86 nmol/L,复合物发出明亮的红色荧光。2017 年,Tan 等[92]筛选获得了另一种可以特异性结合DIR 染料的RNA 适配体DIR2s-Apt,亲和力为966 nmol/L。

Fig. 3 Fluorescent RNAs and fluorophore ligands图3 目前已有荧光RNA适配体及其荧光团配体
基于荧光蛋白的生色团具有小分子质量、电中性、背景荧光弱的特性,研究人员期望获得类似荧光蛋白性能的“拟荧光蛋白RNA”,即用一个RNA适配体去结合、包裹并激活生色团的荧光。目前已报道的荧光RNA 适配体及其配体在光谱上的分布如图3。2011 年,Jaffrey 研究团队[93]模拟GFP 荧光团HBI 合成了染料分子DFHBI 及其RNA 适配体Spinach。DFHBI在没有结合Spinach时的背景荧光很低,结合Spinach 之后相对荧光强度较高,平衡解离常数(KD)为537 nmol/L。由于Spinach 热稳定性差,亮度很低,研究者们相继研究出Spinach2[94]、 Broccoli[95]、 iSpinach[94]和Baby Spinach[96]的RNA 适配体,提高了它们的亮度和热稳定性还降低了其对离子浓度的敏感性并缩小了它的尺寸。然而由于DFHBI 及其类似物极易发生顺/反光异构化,因此以其为荧光团开发的荧光RNA 大多存在较为严重的光猝灭现象。2017 年,科学家们在RFP 的荧光团基础上设计了新的染料分子DFHO。通过体外筛选得到了选择性结合DFHO 的RNA适配体Corn[97]。Corn分子体积小(~30 nt),但其光稳定性优于Spinach 和Broccoli,可以提供高精度的定量结果。这充分说明,光稳定性取决于染料分子与RNA 适配体形成的复合物,而不仅仅取决于染料分子。这些荧光RNA 适配体与染料分子结合后亮度较低,且亲和力较弱、热稳定性较差、容易错误折叠,且Spinach、Broccoli 和Corn不具备生物正交性。
2019年,杨弋课题组与朱麟勇课题组[98]构成的联合攻关团队在荧光RNA 适配体领域取得了突破性进展,他们基于全新的分子设计理念设计合成了全新的荧光团分子HBC。HBC 是结构上具有乙烯桥的分子转子。在自由溶液中,分子转子受激发时获得的能量主要通过围绕乙烯桥旋转运动进行非辐射衰减,因此HBC 在水溶液中几乎没有荧光;当分子转子被蛋白质或核酸结合时,乙烯桥的运动会被限制,受激发时获得的能量会通过产生荧光的方式进行辐射衰减。Pepper是能够特异性结合并激活HBC的RNA适配体。Pepper对HBC有着很强的结合力,KD为3.5 nmol/L,结合后的荧光增强倍数超过3 000 倍。另外,其热稳定性好,光谱涵盖绿色(485 nm)到红色(620 nm)的荧光波段,为实现多种RNA 代谢酶活性检测以及多色检测提供了应用前景。此外,HBC 有着极低的细胞毒性且容易进入细胞,相对于Broccoli和Corn,Pepper在大肠杆菌细胞和哺乳动物细胞中的荧光亮度要高一个数量级以上。因此Pepper 系列荧光RNA 适配体也为活细胞中研究目标RNA 的功能提供了极具价值的工具。
基于荧光RNA 适配体的应用分为体内成像与体外检测两种。基于核酸代谢相关酶的活性研究主要集中在体外的检测,上述荧光RNA 适配体可被用于构建无标记的生物传感器,相较于早期的方法能够实现更加简便、灵活和高灵敏的检测。譬如早期在体外检测转录的方法很大程度上依赖于放射性核苷酸的掺入或基于杂交的方法检测转录过程以及转录酶的活性。近年来,将蛋白质输入直接转导至核酸输出是一种提高蛋白质检测灵敏度的创新策略,因为它可以进一步整合多种核酸扩增技术,如环介导等温扩增(LAMP)、链置换扩增(SDA)、杂交链式反应(HCR)和滚环扩增(RCA)[99-102]等。
2.3.2荧光DNA适配体
相比于荧光RNA适配体,荧光DNA适配体具有更好的稳定性,因此在生物分析和细胞外分子成像中具有更广泛的应用。与荧光RNA 适配体的发展相比,荧光DNA 适配体的发展较为缓慢,基于荧光DNA 适配体构建小分子或蛋白质等生物传感器的报道较少。
很多已知的G 四联体DNA 比如EAD、c-Myc、c-kit2、bcl-2、c-kit1、Kras、T95、TBA、Hras、HTG-21、Telo21、Oxy、Hum 21,如花菁染料、三苯基甲烷染料、咔唑、卟啉等这些荧光染料配体在与这些G 四联体DNA 结合后荧光被显著增强[103]。不同分子与G四联体的结合位点不同,荧光响应机制也不同。根据不同的折叠拓扑结构,G四联体可以识别凹槽中的小分子,并将小分子堆叠在凹槽的顶部或底部上,或者允许小分子插入G四联体的平面之间。静电力、范德华力、氢键、π-π堆积和其他非共价相互作用共同稳定了G四联体与染料配体之间的结合。大多数G四联体的配体都含有一个平面芳香核,其通过π-π堆积与外部G四联体相互作用,侧链通过氢键与DNA 碱基和环的磷酸骨架相互作用后形成稳定的复合物。Jin 等[104]设计并开发了V 型染料BPBC。BPBC 在水性缓冲液条件下几乎没有荧光,与平行G 四联体(EAD、c-Myc、c-kit 2 等)结合后荧光增加330~1 800 倍,与单链或双链DNA结合后荧光仅增加约30倍,与反平行G 四联体结合时增加30~110 倍。G 四联体共有3种结构类型:平行G四联体、反平行G四联体和混合型G四联体,常见G四联体的序列及其结构类型见表4。
除了表4中所列举的一些已报道的G四联体可以与通用的G四联体特异性染料结合后产生荧光激活的DNA外,还有一些是经过严格的SELEX筛选和结构优化而得到的只针对某种染料具有激活效果的特异性荧光DNA 适配体,它们之中有G 四联体也有非G四联体。Sando等[105]于2007年报道了第一个荧光DNA 适配体并命名为Class1-1-mini,其可以与常规的DNA 染料Hoechst 的衍生物产生近200倍的荧光增强。自2007年至今,已有报道的荧光DNA 适配体共有13 条,适配体大小在18~99 nt之间。根据文献的报道把目前已有的荧光DNA 适配体进行了归纳整理[104-116](表5)。在荧光DNA适配体的筛选工作中,裴仁军研究员团队的工作比较突出,在已报道的13条适配体中,有8条出自该团队,分别是MG.1-3、BBR4S3、ThT.2-2、DIR2-1、CV30S、Nm1、Nm2 和ZnP1.2。这些荧光DNA 适配体所使用的染料分别为孔雀石绿、小檗碱、硫磺素T、DIR、结晶紫、N-甲基卟啉二丙酸 IX等常见的核酸染料[110,112-116]。达泊氧磺酰乙二胺(SEDA)和达泊氧磺酸(SA)是达泊氧染料的两种衍生物。DAP-10-42 可以同时激活SEDA 和SA 两种染料分别722倍和35倍,由于其与染料的较高亲和力,常被用来设计为生物传感器[107]。Jaffrey 团队[111]于2022 年报道了一种荧光DNA 适配体适配体Lettuce,可以有100倍左右的激活效果,其激活的染料是DFHBI-1 T,与作为荧光RNA 适配体的Spinach的染料DFHBI相似,都是模拟GFP荧光团HBI的染料分子。聂舟团队[117]报道了一组类似于RFP发色团的全新多色发色团的设计,证明了G四联体可以将DFHBFSI 等RFP 发色团类似物封装入G 四联体形成的空隙空间中,其激发波长跨越了583~668 nm,几乎覆盖了整个RFP 家族的荧光波段。

Table 4 Common sequences of G-quadruplex and their structural types表4 常见的G四联体的序列及其结构类型

Table 5 Fluorescent DNAs and fluorophore ligands表5 目前已报道的荧光DNA适配体及其荧光团配体
本文整理了目前已有的荧光DNA 适配体及其配体在光谱上的分布(图4a)以及各荧光DNA 适配体的二级结构的模拟结果(图4b)。相较于荧光RNA 适配体领域已开发出Spinach、Broccoli 和Pepper 等一批性质优秀的适配体而言,目前荧光DNA适配体的发展仍不足,尚有需要改善的空间,比如进一步开发出碱基数少、分子质量小、高荧光激活倍数且高亲和力的适配体。而且需要注意的是,荧光核酸适配体通常需要镁离子的激活,但是不同的荧光核酸适配体对于镁离子的需求浓度不同,因此开发出低镁离子依赖以及镁离子不依赖的荧光核酸适配体亦是未来的发展需求。

Fig. 4 The spectral distribution (a) and secondary structure (b) of fluorescent DNAs图4 目前已有荧光DNA适配体的光谱分布(a)及二级结构(b)
综上,荧光核酸适配体领域中已有一批荧光DNA适配体和荧光RNA适配体被开发并用作分子生物学研究的新工具,其中一项重要的应用就是目前基于荧光DNA 适配体和荧光RNA 适配体的探针。探针的设计策略主要是将荧光核酸适配体的序列设计在检测体系之中,将目标酶的催化作用与荧光核酸适配体的合成或降解相关联,使作为输出信号的荧光上升或下降,同时可结合纳米技术、核酸扩增技术等实现信号的级联放大,大大提高荧光核酸适配体的检测灵敏度,这对于高通量药物筛选、体外诊断、预后监测等多个领域都具有重要意义。
3 基于荧光法的核酸酶学分析探针的应用
基于荧光法的核酸酶学分析探针在核酸检测、RNA 代谢检测以及各种核酸代谢相关酶的活性分析等多个领域展现出其强大的优势。相关领域的检测会随着荧光法的更新与迭代,不断提高探针检测的灵敏度、特异性和通用性。研究者可以通过加入非特异性核酸染料来实现单色的实时荧光检测(图5a),该方法是有效的,但是不能提供多重靶核酸的即时检测,且具有较高的背景荧光。
基于FRET原理的第二代荧光法通过标记多种颜色的荧光基团,克服了核酸染料不能检测多重核酸的局限。这其中最著名的方法是分子信标法[59]。这种方法可以简单快速地检测靶标序列[72-73](图5b)。然而分子信标法的问题在于它需要至少合成含有两种不同小分子共轭物即荧光染料和猝灭剂的发夹寡核苷酸,且为了准确鉴定靶核酸中的关键序列能特异性地与分子信标杂交而不与检测样品中其他相似序列杂交,需要设置多组多色的分子信标,这需要花费大量的时间和人力,增加了检测成本。

Fig. 5 Schematic diagram of nucleic acid detection based on fluorescence method图5 基于荧光法的核酸检测示意图
基于荧光核酸适配体原理的第三代荧光法对核酸的检测有多种策略,有的研究者们通过对荧光DNA适配体(如Lettuce)和荧光RNA适配体(如Pepper、Spinach、Broccoli)进行修饰,使其在5'和3'端含有侧翼序列,这些序列可与靶核酸序列杂交[94-95,111,118-120]。通过对荧光核酸适配体进行修饰或者将荧光核酸适配体分裂成两个无荧光的RNA,缩短关键螺旋茎降低其热力学稳定性,使荧光核酸适配体无法折叠。当侧翼序列与靶核酸结合时,适配体可以折叠并产生荧光,大大提高了该方法的特异性。也有研究者将荧光核酸适配体技术与等温扩增技术相结合,实现恒温下信号的级联放大实现高特异性和高灵敏度的miRNA 检测。随着技术的不断发展,等温扩增技术已经形成了一个庞大的家族,包括滚环扩增(RCA)、指数扩增反应(EXPAR)、杂交链式反应(HCR)、催化发夹装配(CHA)、链置换扩增(SDA)、双链特异性核酸酶信号扩增(DSNSA) 及其环介导等温扩增(LAMP)等,这些技术的实现主要依赖于酶的聚合作用、水解反应或无酶链替换过程[121-125](图5c)。
荧光法在核酸检测和RNA代谢、DNA代谢相关酶活检测方面发挥着重要作用,为分子生物学和生物医学工程方面提供了重要的支持。接下来,将以T7 RNA 聚合酶、甲基转移酶和T4 多核苷酸激酶等为例,具体讨论三代荧光检测法的区别与联系。
3.1 T7 RNA聚合酶的活性检测
T7 RNA 聚 合 酶(T7 RNA polymerase,T7 RNAP) 可 催 化从5'到3'方 向的RNA 合 成。T7 RNAP已广泛应用于原核生物和真核生物中的高水平基因表达,也被用于RNA干扰、RNA编辑、合成基因电路、构建Vaccinia/T7 混合系统等。T7 RNAP是体内和体外方法中广泛使用的最重要的酶之一,检测T7 RNA聚合酶的活性对于分子生物学和生物工程领域具有重要意义[126]。
3.1.1基于核酸染料法的酶活性检测
SYBR Green II是可以非特异性结合RNA的核酸染料,有研究者使用该染料对T7 RNA聚合酶突变体文库进行高通量筛选:加入修饰的NTP 底物和含有T7启动子的双链DNA模板进行转录,转录后使用DNA 核酸酶I 消化模板,加入RNA 染料SYBR Green II,只有成功转录产生产物的孔会被检测到荧光,最后获得了一个对2-甲基硒修饰的NTP耐受度更高的T7 RNA聚合酶突变体。该方法直观简单,但是具有较高的背景荧光(图6a)。
3.1.2基于FRET原理的荧光探针法的酶活性检测
有研究者使用T7 RNA 聚合酶将荧光rUThioTP残基直接掺入RNA 中,随后用T7 RNA 聚合酶将rUamiTP 残基直接引入RNA 中,然后与亚基丙二腈烯胺(P3)反应。所得到的多重标记RNA在RNA中的两个荧光标记之间表现出FRET,当RNA从单链结构转化为G 四联体时,FRET 信号强度增加。这种使用RNA 聚合酶将位点特异性FRET 标记到RNA中的方法提示了在RNA中的预定位置进行其他不同位点特异性修饰的可能性(图6b,c)。还有研究者利用T7 转录期间从起始到延伸转变过程中的构象变化,使得供体和受体之间的距离发生改变,从而发生荧光的变化。这种方法相比于核酸染料法大大提高了特异性,但是由于需要体外合成或引入荧光团等,操作复杂[126-127]。
3.1.3基于荧光核酸适配体法的酶活性检测
研究者将荧光RNA适配体引入T7 RNAP活性的检测方案,将Broccoli的反向互补序列设计在转录模板中。当T7 RNAP 不存在时,体系中不会有荧光RNA适配体的产生即不会有荧光;当T7 RNA聚合酶存在时,随着转录产物Broccoli 不断产生,荧光信号逐渐增强,并且荧光信号的强弱与T7 RNAP的活性关联。这种方案可以方便地应用于各种噬菌体来源的体外转录系统,并为开发更优的体外转录系统提供平台(图6d)。该方法无需化学合成、体外标记等操作,简化了实验步骤,且由于荧光RNA 适配体的高荧光激活倍数使得该方法的检测灵敏度极高[128]。
3.2 甲基转移酶的活性检测
DNA甲基转移酶(DNA methyltransferase,DNA MTase)能够将甲基S-腺苷甲硫氨酸(S-adenosylmethionine,SAM)迁移到目标腺嘌呤或胞嘧啶残基,从而在DNA 识别中发挥重要作用[129]。
3.2.1基于核酸染料的酶活性检测
研究者结合甲基化特异性和基于SYBR Green的荧光定量PCR 进行O6-甲基鸟嘌呤-DNA 甲基转移 酶 (O6-methylguanine-DNA-methyltransferase,MGMT)启动子甲基化分析。这种技术是一种高度特异性、灵敏且可重复的方法,它不仅可以定量测定完全甲基化的序列,而且可以根据百分比定量测定完全未甲基化的亚硫酸氢盐转化的MGMT DNA 种类。该方法操作复杂,耗时长,且只能检测单重信号[130]。

Fig. 6 Schematic diagram of T7 RNA polymerase activity detection based on fluorescence method图6 基于荧光法的T7 RNA聚合酶活性检测示意图
3.2.2基于FRET原理的荧光探针的酶活性检测
研究者通将分子信标与酶促级联反应相结合,利用甲基转移酶与核酸内切酶(如DpnI 和Nt.AlwI)的序列识别特异性,将限制性内切酶识别位点设计到模板中,在甲基转移酶存在时触发切割效应伴随荧光增强,当甲基转移酶不存在时荧光不增加(图7b)。该方法对于人体无损伤,检测时间短,特异性强,可以设置多重荧光检测[131]。
3.2.3基于荧光核酸适配体的酶活性检测
检测原理与基于FRET的荧光探针法类似,不同点在于,荧光核酸适配体法中无需再添加化学合成的含有荧光团标记的探针,而是利用G四联体形成的DNA 酶与凝血酶的显色反应而实现甲基转移酶活性检测策略(图7a),原理简单,而且该方法中G 四联体序列可以替换为荧光强度更高的NG16、CV30S、ZnP1.2、Apt5.9-32 或者Lettuce 等荧光DNA 适配体,该方法是一种具有通用性的原理简单、操作容易、无标记的检测甲基化酶活性的方法[132]。

Fig. 7 Schematic diagram of methyltransferases activity detection based on fluorescence method图7 基于荧光法的甲基转移酶活性检测示意图
3.3 T4多核苷酸激酶的活性检测
T4 多核苷酸激酶(T4 polynucleotide kinase,T4 PNK)可以促进核酸5'端的磷酸化,参与细胞修复受损的DNA。有研究表示DNA连接酶和多核苷酸激酶的活性与癌细胞的转移相关,因此针对它们的活性研究是很有必要的。
3.3.1基于核酸染料的酶活性检测
研究者于2018年开发出一种使用SYBR Green I核酸染料的DNA 连接酶和磷酸激酶触发的超分支滚环扩增(HRCA)荧光检测平台,在这个体系下如存在T4 PNK便可以将所示探针环化,并作为模板开启HRCA,产生大量双链DNA,其与SYBR Green I结合后荧光显著增强(图8a,b)。
3.3.2基于FRET原理的荧光探针的酶活性检测
研究人员开发了一种一步法、灵敏快速的荧光分析T4 PNK活性的方法,该方法采用环状分子信标荧光探针(图8c)。在T4 PNK 存在的情况下,与探针互补的两个短寡核苷酸中的一个被磷酸化,导致DNA 连接酶促进与另一个寡核苷酸的连接,在连接的DNA 和分子信标之间形成稳定的双链后构成了完整的切刻酶酶切位点,伴随着切刻酶的辅助作用,荧光信号得以产生和富集[133]。同时,研究人员们又开发了一种结合核酸外切酶活性和基于分子信标的T4 PNK活性检测策略,该方法是在赵美萍等[134]首次提出的T4 PNK 酶活性检测方法的基础上进行的改进和优化(图8d)。在这种方法中,发夹探针被T4 PNK 磷酸化,然后立即被λ 外切酶切割,DNA片段得以释放,释放后的DNA片段与分子信标1(MB1)杂交,导致荧光增强。同时,引入分子信标2(MB2)结合在互补域MB1中,导致T4 PNK传感的荧光信号放大[135]。
3.3.3基于荧光核酸适配体的酶活性检测
底物为5'端和3'端均为磷酸基团修饰的单链寡核苷酸,T4 PNK在缺失ATP并且存在ADP的情况下,T4 多聚核苷酸激酶可以显示出磷酸酯酶的活性,可以将3'端的磷酸基团切除留下羟基,在末端脱氧核苷酸转移酶和富含脱氧鸟苷三磷酸的脱氧核苷三磷酸底物存在的情况下引发聚合反应[136],所得延长的DNA 可形成G 四联体,当使用硫黄素T作为G四联体特异性荧光染料时会产生强烈的荧光信号[137](图8e)。
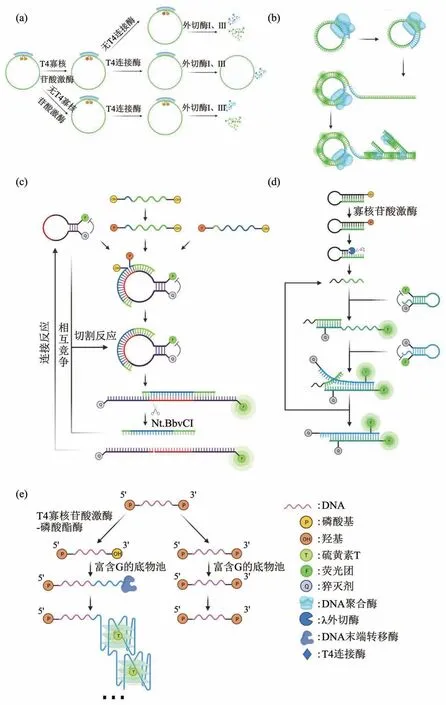
Fig. 8 Schematic diagram of T4 polynucleotide kinase activity detection based on fluorescence method图8 基于荧光法的T4 多核苷酸激酶活性检测示意图
3.4 脱嘌呤/脱嘧啶核酸内切酶1的活性检测
脱嘌呤/脱嘧啶核酸内切酶1 (apurinic/apyrimidinic endonuclease 1,APE1),又称氧化还原因子1(Ref-1),是人体必需的基因调控蛋白和DNA 修复蛋白[138]。作为最重要的DNA 修复酶,APE1 作用于双链DNA(dsDNA)中的脱嘌呤/脱嘧啶位点(AP位点)并启动碱基切除修复(BER)通路[139-140]。APE1 还可以通过其氧化还原功能调节许多转录因子的DNA结合活性。以往研究表明,人体血液中APE1含量的异常变化和组织细胞与多种疾病有关,例如前列腺癌[141]、肺癌[142]、卵巢癌[143-144]和膀胱癌[145]。因此开发出有效检测APE1的酶活性方法对于癌症诊断、预后监测和抗癌药物筛选均具有重要意义。
3.4.1基于FRET原理的荧光探针法的酶活性检测
研究人员们构建了一种含有硫代磷酸化修饰的AP 位点的DNA 探针[146],其在AP 位点3'端的第4个碱基和AP 位点5'端的第5 个碱基分别标记猝灭基团和荧光基团,当不存在APE1 酶时,基于FRET 原理,荧光被猝灭基团猝灭,在APE1 存在的情况下,AP位点的5'端的脱氧核糖-磷酸主链将被快速切割裂解,含有荧光团的短链被释放从而可以检测到荧光(图9a)。该方法无需额外的清理或预处理步骤,简单快速,一步完成,线性工作范围为0.1~5.0 U/ml,检测下限为0.1 U/ml。该方法的灵敏度有限。
随后有研究人员,优化设计了新的检测探针,开发出TdT和Endo IV辅助的双信号放大测酶活力的方法(图9b)。他们设计了3'端进行氨基修饰含有AP 位点发夹状的DNA,当APE1 存在时,其可以切除发夹DNA 底物上的AP 位点,生成3'-OH端,此时TdT可以在3'端继续延伸,通过添加三磷酸脱氧腺苷(dATP)使得底物产生大量的poly-A尾。此时,含有荧光基团和猝灭基团的poly-T探针(猝灭状态)可以大量地杂交在poly-A尾。随后高温加热将APE1 和TdT 失活并加入内切酶IV(Endo IV)来切割AP 位点,产生带有荧光基团的短链,随着其与猝灭基团的分离而发出荧光。当体系中不存在APE1 时,3'端的氨基修饰阻断了末端脱氧核苷酸转移酶(TdT)的延伸,后续反应不发生。该方法具有高灵敏度和高选择性,该方法的检测下限为1.7×10-3U/L。
3.4.2基于荧光核酸适配体的酶活性检测
研究人员开发出基于聚合酶和切刻酶共同辅助的等温扩增方法来检测APE1酶的活性。在发夹结构探针中设计AP 位点,当APE1 存在时,产生3'-OH 端此时在聚合酶克列诺片段(Klenow Fragment)的作用下将模板的发夹结构打开,聚合成双链DNA,产生了切刻酶的酶切位点,伴随着切刻酶与聚合酶的作用下,促进G四联体短链的指数生成,G四联体可以与NMM结合发出荧光(图9c)。该方法具有高灵敏度,检出限为6 U/L[147]。但是在此方法中没有测试其他非特异性核酸内切酶或核酸外切酶的干扰。
最近,研究人员们开发了一种基于滚环扩增结合G四联体的高灵敏度和无标记地检测APE1的新方法。标记有AP 位点的发夹探针可以被APE1 识别并切割,导致引物序列的释放从而启动RCA 反应(图9d)。RCA的模板上含有G四联体的反向互补序列,因此反应中不断产生含有串联的G四联体结构的长链扩增产物,产物可与硫黄素T(ThT)结合产生荧光,实现APE1 的高灵敏度无标记检测。该方法的检出限低至1.52×10-3U/L。
3.5 端粒酶的活性检测
端粒酶是一种逆转录酶,其本身是一种大型核糖核蛋白复合物,负责在线性染色体的3'端逐步合成端粒DNA重复序列(TTAGGG),从而逆转每一轮复制中的DNA 丢失[148]。且最近的研究报道,除了维持端粒外,端粒酶还参与基因表达调控、细胞增殖、细胞凋亡、WNT/β连环蛋白信号、NF-κB信号、MYC 驱动的肿瘤发生、DDR、细胞黏附和迁移、上皮-间质转化等活动[149-152],这些端粒酶的功能与肿瘤的发生过程有重要联系。因此,作为一种通用的肿瘤生物标志物,研究端粒酶的活性和抑制作用对于癌症的诊断和治疗具有重要意义。
3.5.1基于核酸染料法的酶活性检测
传统的TRAP测定需要进行聚丙烯酰胺凝胶电泳和放射自显影,以此使典型的六碱基产物阶梯可视化,并在光密度测定后定量端粒酶活性。研究者结合了SYBR Green的荧光定量PCR与传统的端粒重复扩增方案(TRAP),优化了实时定量的TRAP测定[153],但其应用受到扩增相关的错误和耗时程序的限制。
3.5.2基于FRET原理的荧光探针法的酶活性检测
为了解决基于PCR 的局限性,双扩增荧光测定法被用于无PCR检测端粒酶活性[154]。检测平台应用了一种拱形结构DNA 探针来专门控制链置换反应和随后的酶辅助扩增。端粒酶底物引物被端粒酶延伸形成含有多个TTAGGG 重复单元的长延伸产物。因此,一个延伸产物可以通过链置换反应释放出多个触发DNA(t-DNA),从而实现第一次扩增。随后,t-DNA特异性地打开分子信标以恢复荧光。同时,t-DNA 在切口核酸内切酶的帮助下循环,不断打开越来越多的分子信标,实现二次扩增。该方法能够测定相当于5 个HeLa 细胞或10 个CCRF-CEM细胞的端粒酶活性。
3.5.3基于荧光核酸的酶活性检测
研究团队研发了“DNA机器”,它是一款由T7核酸外切酶(T7 Exo)、无标记识别分子信标(RMB)和具有突出5'端的信号分子信标(SMB)组成的探针,用于端粒酶活性的灵敏检测[155]。首先,端粒酶延长端粒酶底物(TS)引物,产生具有串联重复序列(TTAGGG)n的端粒酶延长产物(TEP)。EP通过与RMB杂交激活DNA机器,展开具有凹陷 5'端的 RMB,使RMB 从T7 Exo 脱出,发生T7 Exo 辅助循环切割,从而释放完整的TEP和大量DNA 片段(触发DNA)。随后,触发DNA特异性打开SMB 并被T7 Exo 回收,释放出多个G 四联体(G4)结构。最后,TEP 和释放的G4 结构与N-甲基-中卟啉IX(NMM)强烈相互作用后产生显著增强的荧光。以这种方式,每个端粒酶介导的延伸事件被有效地转化为放大的荧光信号。该检测方式能够测量相当于50 个HeLa 细胞/ml 的端粒酶活性,线性范围为50~2 000 个细胞/ml,相较其他方式大大提高了灵敏度。
3.6 8-羟基鸟嘌呤DNA糖基化酶的活性检测
由ROS 促成的氧化产物8-羟基鸟嘌呤(8-oxoG)是DNA 中发现的最普遍的碱基损伤。8-羟基鸟嘌呤DNA 糖基化酶(OGG)是一种关键的碱基切除修复(BER)酶,可以识别8-oxoG 并从DNA 中切除[156]。OGG 的表达水平与多种人类癌症密切相关,包括肺癌、胃癌、胆囊癌和膀胱癌[157-160]。
3.6.1基于核酸染料法的酶活性检测
研究者开发了一种简单的混合读取测定法,使用多重循环酶促修复扩增来灵敏检测hOGG1[161]。hOGG1 活性产生一个AP 位点,AP 位点会被脱嘌呤/脱嘧啶核酸内切酶1(APE1)切割,产生带有游离基团的底物片段3'-OH端。底物片段可以启动循环酶促修复扩增,指数级地产生触发器,并使用SYBR Green II 作为荧光染料可以简单地检测扩增产物。该方法可以灵敏地测量hOGG1,检测限为2.97×10-2U/L。
3.6.2基于FRET原理的荧光探针法的酶活性检测
研究者开发了一种基于自主核酸外切酶III(Exo III)辅助信号放大的酶活性检测技术,构建了用于hOGG1 活性检测的新型高灵敏度荧光生物传感平台[162]。发卡探针HP1在8-oxoG位点被切割并被Exo III 消化,释放触发DNA 片段(tDNA1)。tDNA1 与发卡探针HP2 部分杂交,启动Exo III 循环切割,释放另一个触发DNA片段(tDNA2),进而触发DNA 荧光探针(FP)的循环切割,从而释放大量荧光信号,用于hOGG1 活性检测。基于荧光探针的酶活检测具有高灵敏度以及良好的选择性,直接测量检测限低至1 U/L,并具有等温实验条件、简单和方便的优点。
3.6.3基于荧光核酸的酶活性检测
研发团队报道了一种利用λ外切核酸酶切割进行DNA 糖基化酶活性测定的方法[163]。hOGG1 选择性地切割含有8-oxoG的DNA双链体时,能够生成具有5'-PO4端的新DNA双链体,从而被λ外切核酸酶消化,释放出游离的G四联体单链,并与血红素结合形成具有催化活性的G 四联体-血红素DNAzyme。DNAzyme可以催化形成ABTS-,因此hOGG1的活性能够通过紫外-可见光指示吸收强度来进行灵敏的定量测定,线性范围从0.05~32 U/ml,检测限0.01 U/ml。
3.7 尿嘧啶DNA糖基化酶的活性检测
尿嘧啶DNA 糖基化酶(UDG)是一种高度保守的损伤修复蛋白,能够通过糖基化酶活性启动碱基切除修复途径,切除尿嘧啶碱基[164]。UDG在维持细胞周期调控、细胞凋亡、病毒增殖等基因组完整性方面起着关键作用[165]。DNA糖基化酶的异常表达与人类多种疾病密切相关,如人类免疫缺陷、布卢姆综合症、癌症等[166-169]。因此,UDG的检测对于研究许多基本生化过程的机制和功能以及疾病诊断尤其重要。
3.7.1基于核酸染料法的酶活性检测
在众多基于核酸染料法中,研究者们为更精确的UDG 检测方式设计了各种核酸扩增的方式,包括杂交链式反应(HCR)[170]、催化发夹组装(CHA)[171]、指数扩增反应(EXPAR)[172]、链置换扩增(SDA)[173-174]和滚环扩增(RCA)[165,175-176]。但这些方法往往伴随着复杂的探针设计和反应步骤。其中,自启动多重RCA 法获得的检测灵敏性较高[165],线性范围为0.05~1.25 U/L,检出限为1.7×10-2U/L。
3.7.2基于FRET原理的荧光探针法的酶活性检测
多个研究团队设计了利用分子信标检测尿嘧啶-DNA糖基化酶(UDG)的策略。2007年时,就有研究团队利用FRET原理检测UDG酶活[177]。双链探针由P1 和P2 两条反向互补链组成:一条标记DABCYL 作为受体,另一条标记TAMRA 作为供体,中间位置有四个尿嘧啶碱基。当UDG 添加至体系中时,具有4个AP位点的P1P2探针因熔化温度降低而被分离,从而发出荧光信号,低检测限为33 U/L。虽然这种检测方式较传统方法在灵敏度与简便性上有所突破,但依旧受限于复杂环境下的不稳定性。
2014 年,研究者报道了一种基于尿嘧啶修饰的分子信标,探针的茎中仅包含两个尿嘧啶碱基(6个碱基对),非常适合作为UDG的底物[178]。尿嘧啶被切除后,产生的AP位点将显着降低分子信标的熔化温度,并且由此产生的荧光团标记信标与互补链解离,使得荧光显着增加。该方法无需扩增,低检测限为5 U/L。
3.7.3基于荧光核酸的酶活性检测
研发团队利用G四联体DNAzyme链(GS)被UDG活性激活的设计,开发了一种比色测定UDG活性的无酶和无标记策略[179]。该策略依赖于目标激活的立足点介导的链置换(TMSD)电路,采用由含尿嘧啶链(US)和催化剂链(CS)组成的检测双链探针。样品中存在的UDG会切割US内的尿嘧啶碱基并破坏检测双工探针的稳定性,释放CS。游离CS 促进TMSD 反应,从而释放出最初被阻断链(BS)笼罩的G 四联体DNAzyme 链(GS)。大量GS 被UDG 活性激活,并且通过释放的GS 的过氧化物酶模拟活性促进的ABTS氧化产生明显的比色信号。基于这一设计原则,能够非常灵敏且选择性极佳地检测到UDG 活性。通过可靠地测定人血清中的UDG 活性也证明了该测定的实际适用性。该方法的检测限低至6 U/L。
综上,无论是基于荧光RNA 适配体的酶活性检测方法还是基于荧光DNA 适配体的酶活性检测方法,其本质都是一样的,即利用荧光适配体实现高特异性、高灵敏度、无标记的实时荧光定量检测酶的活性,且上述体系由于其原理简单、合成容易、成本低廉、方法特异性强、检测灵敏等优点,相较于非特异性的核酸染料法以及TaqMan 探针法展现出巨大的优势。因此,基于荧光核酸适配体的方法具备用于新型高通量药物筛选和临床即时诊断的高通量分析的潜力和实力。
4 总结与展望
本文从比色法、高效液相色谱法、放射性标记法、凝胶电泳法、电化学法、酶联免疫吸附法和聚合酶链式反应法等传统酶活性检测方法讲起,分别比较了各个方法的优点与缺点。目前这些方法具有操作复杂、对仪器的要求高、具备放射性危害、灵敏度低等限制性因素,很难进行高通量、高灵敏度、高特异性以及低成本的检测。本文将荧光法按照其发展历史分为荧光染料法、基于FRET原理的特异性探针法和荧光核酸适配体法(表6),把这3种荧光检测方法的优势、劣势、代表性研究以及作用原理进行了整理。

Table 6 Comparison of three fluorescence methods表6 三种荧光法比较
基于遗传编码的荧光核酸适配体检测方法的最大优势就是其原理简单、成本低廉、特异性高、稳定性强、安全性好以及易于实现高通量等优势。荧光核酸适配体将会在接下来的时间中,逐渐接过核酸染料和TaqMan 探针的接力棒,为酶活性检测这一领域开展新的篇章。荧光核酸适配体法在酶学分析与检测上有着前所未有的热度,不同于无法穿过细胞膜的核酸染料、需要体外合成的TaqMan 等探针,分子质量小、设计灵活的遗传编码的荧光核酸适配体可以在活细胞内进行高时空分辨率的成像,直观地展现活细胞内核酸代谢的精确位置和数量,为核酸代谢相关酶的体内细胞环境的功能研究提供更好的机遇。
目前来说,荧光RNA 适配体的发展已取得了长足的进步,并在活细胞成像领域展现出巨大潜力,评价荧光RNA 适配体的性质需要从染料背景荧光、光稳定性、亲和力以及生物正交性等多方面进行考察,比如荧光RNA 适配体Mango 和Pepper具有较高的亲和力(纳摩尔级别)。然而,已有报道的生物传感器大多基于早期开发的荧光RNA 适配体,且目前基于RNA 相关酶的酶活性检测方法受限于针对酶作用机制的研究。荧光核酸适配体目前在应用过程中存在的不足有以下几点。
首先,目前在各个领域,包括酶活检测领域应用较为广泛的荧光DNA 适配体均为G 四联体,检测时加入NMM、TO等G四联体的通用染料。G四联体是由Hoogsteen氢键连接4个G形成环状平面,两层或以上的四分体通过π-π堆积形成四联体,因此链与链之间存在相互作用,当把不同种类的G四联体放在同一体系中,极易发生分子之间的错误交联。这样的错误交联一旦发生,一个是无法形成正确的G四联体结构也就无法检测到荧光。另外,目前没有只针对某种G四联体染料响应的序列,大多数的染料均为G四联体结构依赖型的通用染料,例如研究者们开发出的荧光DNA 适配体DAP-10-42是基于dapoxyl SEDA 筛选得到的,但是有研究证明它对多种芳基甲烷类染料均有激活效果,其中对于AO[107]的激活效果达2 070倍,激活倍数高于其筛选的靶标染料(722 倍)。基于上述两个原因,当前已有的荧光DNA适配体很难实现生物正交性,即同一体系中很难存在2 个及以上的不同的G 四联体。
其次,应用荧光核酸适配体设计的各种探针,在其基于荧光核酸适配体的信号输出阶段需要依赖荧光核酸适配体的正确折叠。荧光核酸适配体发挥功能需要其形成特定的二级结构,如果二级结构无法正常形成,那么荧光核酸适配体不能折叠成具有激活染料功能的二级结构,则无法产生荧光。Baldrich 等[180]探究了基于适配体的生物传感器在不同的金属离子浓度、不同温度等条件下受到的影响。因此,如果溶液中离子浓度、温度、pH 等环境不能满足荧光核酸适配体的要求,则荧光核酸适配体无法正常折叠,最终会影响荧光强度的检测。
最后,高性能的荧光核酸适配体开发难度极大,这是目前荧光核酸适配体的应用受限的一大原因。目前已报道的荧光核酸适配体,尤其是荧光DNA 适配体,其与染料配体结合后的荧光亮度较低、亲和力较弱等自身固有的性质仍待改进[104-116]。研究者们应用荧光核酸适配体进行检测、分析的过程中,会发现其检测限受到极大的限制,虽原理简单操作方便,但是基于目前已报道的荧光核酸适配体很难实现单分子检测。
基于上述,本文对于荧光核酸适配体的发展进行如下展望:a. 当前的荧光核酸适配体筛选的方法较为传统而且周期长,因此需要优化荧光核酸适配体的筛选策略,建立周期短、通用性强的筛选平台; b. 目前基于荧光核酸适配体的生物探针主要以“分裂式”、“二聚体式”、“倒置融合”和“环状排序”等形式进行设计检测microRNA、肿瘤标记物等,需加强基于荧光核酸适配体的生物探针的开发力度;c. 开发基于荧光核酸适配体的活细胞上高时空分辨率的成像,直观地展现活细胞内核酸代谢的精确位置和数量,为核酸代谢相关酶的体内细胞环境的功能研究贡献更多策略;d. 目前荧光核酸适配体尤其是荧光RNA 适配体已经基本覆盖可见光光谱的各个波段,但是近红外、远红外波段上的荧光DNA适配体和荧光RNA适配体数量较少,应该拓展当前荧光核酸适配体的光谱波长,丰富荧光核酸适配体的种类和数量以及正交型荧光核酸适配体对,实现多重检测与疾病诊断;e. 针对酶的作用机制研究需要更加深而广,才能更加有助于研究者们基于不同种类的酶的特有性质设计针对性更强的酶活性检测体系,从而搭建基于荧光核酸适配体的核酸代谢新型药物筛选平台;f. 荧光DNA 适配体相较于荧光RNA 适配体具有更加稳定的物理化学性质,因此荧光DNA 适配体未来将在成为医学诊断和预后诊断中扮演更加重要的角色。
荧光核酸适配体的开发仍然在不断进行,未来核酸适配体有望在生物化学分析、优势酶的定向进化、活细胞成像、医学检验等领域为实验人员提供更广阔稳定的平台。研究者们将进一步丰富荧光核酸适配体的工具库,拓展其在生命科学相关领域内的应用。
