线粒体蛋白CHCHD10与神经退行性疾病*
2023-05-16王腈朱笠
王 腈 朱 笠
(中国科学院生物物理研究所,脑与认知科学国家重点实验室,北京 100101)
线粒体是细胞的能量工厂和信号中转站,其主要功能除了生产ATP 外,还参与氨基酸和脂类等代谢物的生物合成、细胞信号转导、活性氧(reactive oxygen species,ROS)生成、蛋白质稳态维持、代谢调节、免疫反应和调控细胞程序性死亡等[1]。线粒体含有自身的基因组(mtDNA),该基因组含有16 596 个碱基对组成的37 个基因,编码22 个tRNA、2 个rRNA 和13 条多肽链,这13 条多肽链分别是不同呼吸链复合物的组分,参与线粒体氧化磷酸化 (oxidative phosphorylation,OXPHOS)[2-3]。然而,人类细胞的线粒体含有1 200 多种蛋白质,约98%的线粒体蛋白是由核基因组(nDNA)编码的[4]。与大多数线粒体蛋白相似, CHCHD10 (coiled-coil-helix-coiled-coil-helix domain containing 10)由核基因编码,在细胞质中合成,经线粒体蛋白转运系统进入线粒体,定位在线粒体膜间隙 (intermembrane space of mitochondria,IMS)和线粒体嵴上[5]。
诸多研究表明,线粒体稳态失衡与额颞叶变性病 (frontal temporal lobar degeneration,FTLD;也叫额颞叶痴呆(frontotemporal dementia,FTD))、肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)、帕金森病(Parkinson's disease,PD)、阿尔茨海默病(Alzheimer's disease,AD)等多种神经退行性疾病密切相关[6]。在一个线粒体里,mtDNA 通常以多个拷贝存在,而且一个细胞中含有成百上千个线粒体。由于细胞线粒体处于分裂和融合的动态变化中,其数目也是变化的。此外,mtDNA 组更易发生突变,其突变率比nDNA 高100~1 000 倍。因此,线粒体稳态失衡主要包括:a. mtDNA 拷贝数明显减少或mtDNA 缺失产生的mtDNA不稳定性增强,可能导致mtDNA损耗综合征[7-8];b. 线粒体分裂融合动态失衡导致线粒体结构形态改变[9];c. ROS 水平增加[7-8];d. OXPHOS水平改变[7-8]。
运用生物信息学方法预测CHCHD10与线粒体OXPHOS 有关,并通过实验证明其参与线粒体呼吸链复合物COX 活化[10]。研究发现,CHCHD10的点突变S59L 会导致常染色体显性遗传病,表现出运动神经元损伤、认知功能下降等症状[5]。随后,CHCHD10的功能研究备受关注,而且更多与神经退行性疾病相关的CHCHD10 基因突变被发现。本文总结了近年来已发表的关于CHCHD10结构及其线粒体功能的文章,为研究与CHCHD10相关的神经退行性疾病提供新思路。
1 CHCHD10蛋白的一级结构
CHCHD10基因位于人类第22号染色体,其编码的蛋白质由142 个氨基酸组成。CHCHD10 蛋白N 端的前16 个氨基酸是线粒体定位序列(mitochondrial targeting sequence,MTS),C 端99~140 位氨基酸组成其CHCH 结构域(coiled-coilhelix-coiled-coil-helix domain),其中C102和C132,C112和C122分别形成两对二硫键(图1)。
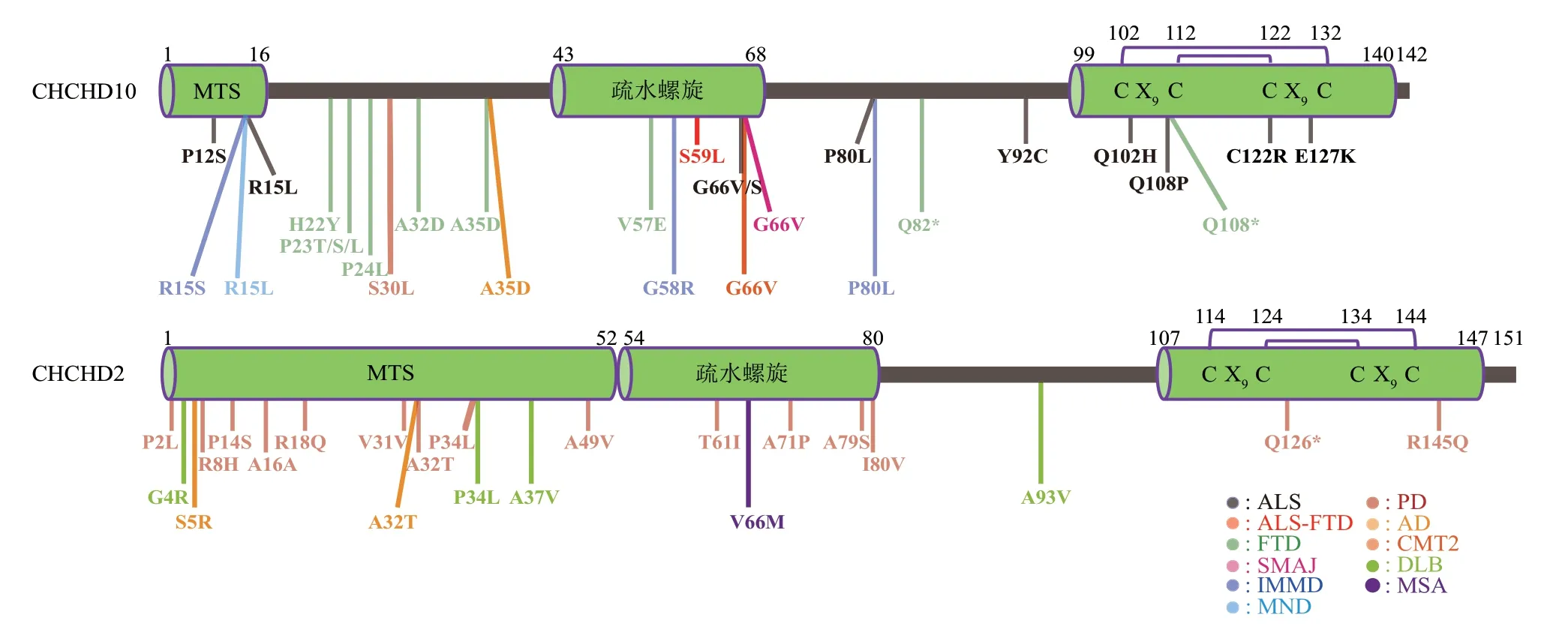
Fig. 1 Primary structures of CHCHD10 and CHCHD2 proteins as well as the amino acid positions of various neurodegenerative diseases associated mutations图1 CHCHD10和CHCHD2蛋白的一级结构及其基因在FTD、ALS和AD等神经退行性疾病中突变位点对应的氨基酸位置
由于二硫键的存在,CHCHD10 通过Mia40/Erv通路进入线粒体,最终定位在线粒体膜间隙[5](图2)。在HeLa细胞中,与对照相比,敲减Mia40后,进入线粒体的CHCHD10 明显减少。过表达Mia40,ALS 相关突变体Q108P-CHCHD10 更多地进入线粒体[11]。CHCH结构域在CHCHD10进入线粒体时发挥作用。删除CHCH结构域或表达CHCH结构域的疾病相关突变体(如Q108P、C122R),进入线粒体的CHCHD10 显著减少。然而,删除CHCHD10的N端1~16这一段氨基酸组成的线粒体定位序列对其进入线粒体无明显影响[11]。但是,不同实验室的研究结果存在差异,Burstein 等[12]发现,单独删除N 端1~16 氨基酸序列或者单独删除CHCH 结构域,均会影响CHCHD10 进入线粒体。
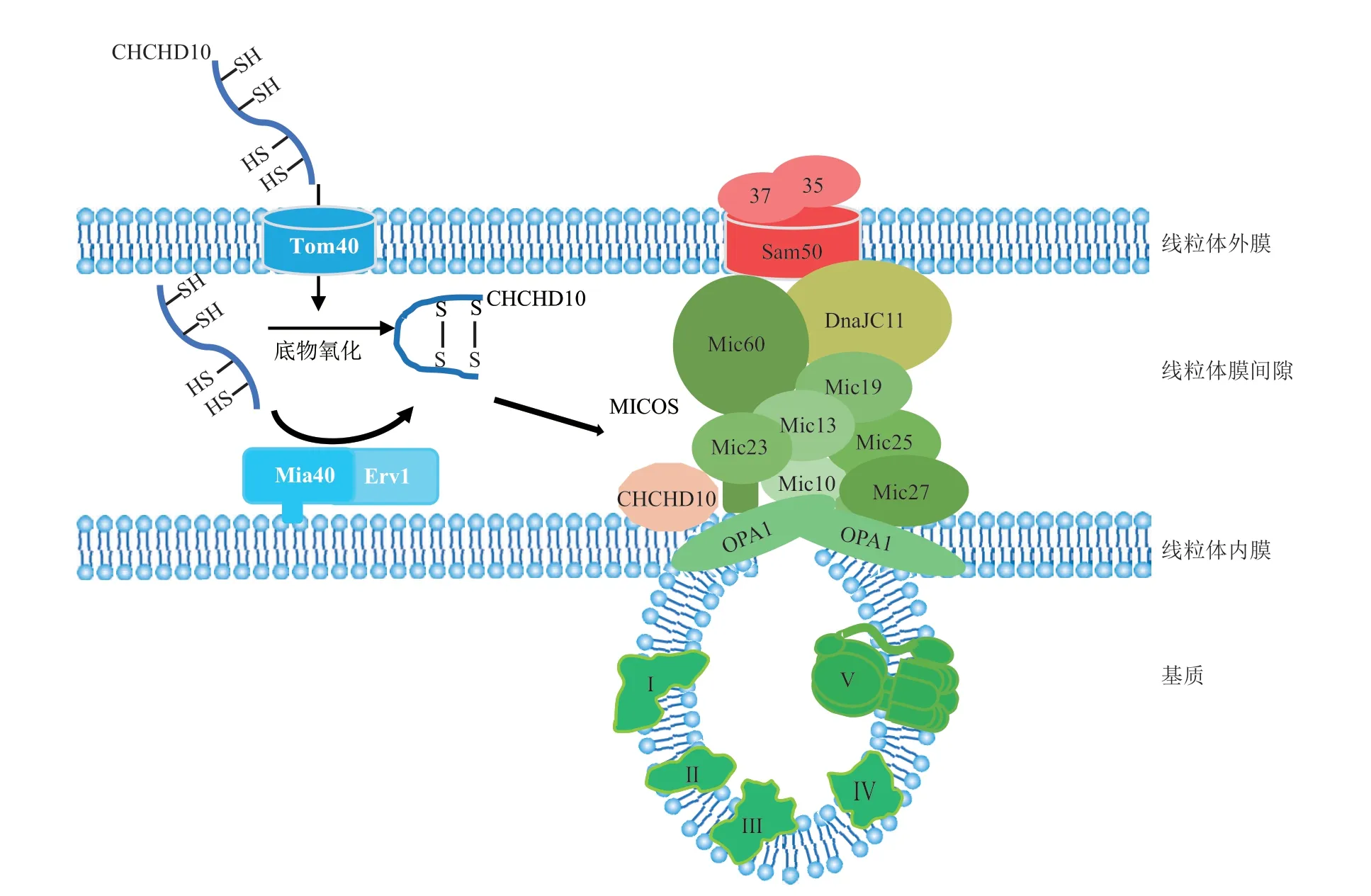
Fig. 2 CHCHD10 is imported into mitochondria through Mia40/Erv pathway and involved in the maintenance of the mitochondrial integrity图2 CHCHD10通过Mia40/Erv途径进入线粒体并参与维持线粒体结构的完整性
2 CHCHD10疾病相关突变体
2014 年,科研人员首次在ALS-FTD 患者的病理组织中筛选到CHCHD10 的突变体S59L[5]。随后更多与神经退行性疾病相关的CHCHD10突变体被报 道, 包括与ALS 或ALS-FTD 相关的p.P12S[15]、p.R15L[16-18]、p.S59L[5,19]、p.G66V[16]、p.G66S[11]、p.P80L[18,20]、p.Y92C[21]、p.Q102H[21]、p.Q108P[11]、p.C122R[11]、p.E127K[11],与FTD相关的p.H22Y[22]、p.P23S[22]、p.P23L[23]、p.P23T[18]、p.A32D[22]、p.A35D[18]、p.V57E[22]、p.Q82*[15]、p.Q108*[24];与常染色体显性运动神经元疾病(autosomal dominant motor neuron disease,MND)相关的p.R15L[25],与常染色体显性线粒体肌病(autosomal dominant mitochondrial myopathy,IMMD)相关的p.R15S[26]、p.G58R[26]、p.P80L[20],与迟发性脊肌萎缩症(late-onset spinal muscular atrophy,SMAJ)相关的p.G66V[27],与2 型腓骨肌萎缩症(Charcot-Marie-Tooth 2,CMT2)相关的p.G66V[28],与散发性PD相关的p.S30L[29],与AD相关的p.A35D[30],等等(图1)。
3 CHCHD10与同源基因CHCHD2
CHCHD2与CHCHD10同属于CX9C蛋白家族,二者的蛋白质序列同源性高达58%[31]。CHCHD10与CHCHD2 存在相互作用,并且与线粒体相关蛋白形成分子质量约为220 ku 的复合物[12,32]。通过受激发射损耗荧光显微术(stimulated emission depletion,STED)观察到CHCHD10 与CHCHD2沿着线粒体形成轨道样结构[33]。在线粒体中,CHCHD10 作为支架蛋白,辅助ARG 激酶(ABL proto-oncogene 2, nonreceptor tyrosine kinase,ABL2)调节CHCHD2 的磷酸化,进而活化COX复合物[31]。CHCHD2 在细胞内可单独形成二聚体或与CHCHD10 形成异二聚体,但是CHCHD10 需与CHCHD2结合才能形成聚合物[34]。
4 CHCHD10的功能
4.1 CHCHD10与线粒体结构完整性
MICOS (mitochondrial contact site and cristae organizing system)复合物位于线粒体内膜上,对线粒体嵴结构的形成和维持至关重要[14],MICOS复合物组装发生障碍会影响线粒体嵴结构的完整性。CHCHD10主要定位在线粒体膜间隙中。免疫电镜显示CHCHD10在线粒体富集[5,34],免疫共沉淀检测到CHCHD10 与MICOS 复合物的Mic60/mitofilin 存在相互作用,因此Genin 等[35]认为CHCHD10 是MICOS 复合物的组成部分。然而,后续其他研究均未能重复出上述结果,即未检测到CHCHD10 与MICOS 复合物中Mic60/mitofilin 之间的相互作用。CHCHD10 蛋白单体分子质量为14 ku。一维的天然胶(或非变性胶)聚丙烯酰胺凝胶电泳 (blue native polyacrylamide gel electrophoresis,BN-PAGE) 显 示,CHCHD10 在170 ku和220 ku与其他蛋白质形成复合物,但在约600~720 ku处没有检测到CHCHD10存在[32]。免疫共沉淀或二维SDS聚丙烯酰胺凝胶电泳(2D-SDSPAGE)共迁移实验,均未检测到CHCHD10 或CHCHD2 与MICOS 复合物的Mic60 或Mic19 存在相互作用。用Mic60 或Mic19 抗体进行免疫共沉淀,然后进行质谱分析,均未检测到CHCHD10或CHCHD2[32]。在HEK293细胞中转染带FLAG标签的CHCHD10,用FLAG 抗体进行免疫共沉淀,可检测到mitofilin,但Mic60/mitofilin 与IgG 也存在相互作用。相反,用Mic60/mitofilin 抗体结合的免疫共沉淀实验,未检测到CHCHD10。因此,Mic60/mitofilin 与CHCHD10 之间可能是非特异性的相互作用[12,33]。因 此,CHCHD10 是 否 是MICOS复合物的必要组成部分,目前仍存在争议。
另外,CHCHD10 对MICOS 复合物的表达影响甚微。在ALS 相关突变体G66V-CHCHD10 患者的成纤维细胞中,MICOS 复合物中mitofilin 表达无明显改变[36]。HeLa 细胞中,敲除CHCHD10 或CHCHD2,MICOS 复合物的组成部分Mic60/mitofilin、Mic19/CHCHD3 和Mic25/CHCHD6 均无明显改变。CHCHD2-CHCHD10 双基因敲除后,MICOS 复合物组成部分Mic60、Mic19/CHCHD3、Mic25/CHCHD6略微减少[34]。
虽然CHCHD10 是否属于MICOS 复合物的组成部分存在争议,但毫无疑问,CHCHD10对线粒体形态结构的完整性维持尤为重要。CHCHD10缺失或者突变,均会影响线粒体嵴结构的完整性。单独敲减CHCHD10或CHCHD10-CHCHD2双基因敲除后,线粒体嵴结构出现异常[33-34,37]。在ALS 相关突变体S59L-CHCHD10 患者的成纤维细胞中,出现线粒体嵴缺失、线粒体呼吸链缺乏、mtDNA损伤修复障碍等异常[5,35]。在HeLa 细胞系中过表达ALS 相关突变体S59L-CHCHD10,或在S59LCHCHD10的转基因小鼠模型中,线粒体嵴均是受损的[5,38-40]。不过,与上述结果不一致的是,携带另外一种ALS 相关突变G66V-CHCHD10 的患者,其成纤维细胞中的线粒体嵴结构无明显变化[36]。目前的研究结果显示,CHCHD10参与维持细胞线粒体的形态结构,但具体以何种方式参与其中尚不明确。
4.2 CHCHD10与线粒体分裂融合
线粒体通过不断的分裂(fission) 和融合(fusion)以维持其正常结构和生理功能,一旦分裂和融合失去平衡将直接导致线粒体形态改变和功能受损。ALS 相关突变S59L-CHCHD10 患者的成纤维细胞中,转染线粒体mitoPAGFP 质粒,在405 nm 激发光条件下连续观察60 min,线粒体荧光强度减弱的速率与对照相比无明显差异,提示S59L 突变对线粒体融合无显著影响[5,35]。使用不同的方法进行实验,得到的结果略有差异。在NIH-3T3 细胞中,同时转染野生型或突变体CHCHD10 (如R15L、S59L) 和mito-dendra2 质粒,48 h 后在405 nm 激发光条件下连续观察线粒体的荧光强度,发现转染突变体的细胞中其荧光强度的改变速率显著慢于对照组,提示线粒体融合受损[41]。虽然CHCHD10 突变对线粒体融合的影响因实验方法不同而存在差异,但已报道的ALS 相关突变(包括S59L、R15S、G58R、G66V)在患者样本、小鼠模型或细胞模型中,均观察到线粒体发生片段化[5,26,36-40]。线粒体动力蛋白样GTP 酶(mitochondrial dynamin-like GTPase)OPA1(optic atrophy 1)是调节线粒体分裂和融合的重要蛋白质。在YME1 样ATP 酶(YME1 like 1 ATPase,YME1L1)和锌金属肽酶OMA1 (OMA1 zinc metallopeptidase,OMA1)这两种蛋白水解酶的作用下,OPA1会形成长、短两种形式,L-OPA1参与线粒体融合过程,S-OPA1 与线粒体分裂相关,L-OPA1与S-OPA1比例失衡将影响线粒体分裂和融合,最终导致线粒体结构异常[37,40-41]。CHCHD10突变体使线粒体片段化或许与OPA1水解相关,但是这一推测需要实验进一步证实。
4.3 CHCHD10与线粒体OXPHOS
OXPHOS 主要发生在线粒体内膜上,由一系列蛋白质复合物完成。糖、脂、蛋白质这三大类营养物质进入有机体后,首先被消化为小分子物质,它们被吸收进入细胞后,通过各种酶促反应形成乙酰辅酶A 等中间产物,然后进入线粒体三羧酸循环,最终生成CO2和H2O。在这一氧化过程中释放的能量,通过线粒体呼吸链复合物供给ADP 与无机磷酸盐,合成能量分子ATP,体内95%的ATP都是通过线粒体的OXPHOS 合成的[42]。运用生物信息学方法预测,CHCHD10是核基因编码的线粒体蛋白,同COX19 一样含有CHCH 结构域,而COX19 参与呼吸链复合物IV(complex IV)的形成,提 示CHCHD10 可 能 与complex IV 有 关[10]。在HeLa 细胞中,通过免疫荧光观察到CHCHD10与线粒体存在明显共定位,且CHCHD10主要定位在线粒体中,这一实验证实了预测的结果[10]。而且,在HeLa 细胞中敲减CHCHD10,经线粒体OXPHOS解偶联剂 (carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone,FCCP)或寡霉素处理,或用含半乳糖的培养基培养细胞,细胞ATP 合成减少,Complex IV 活性降低[10]。随后更多的实验结果表明,CHCHD10 与线粒体OXPHOS水平密切相关。CHCHD10基因缺失的细胞系或携带CHCHD10 突变(如S59L、R15S、G58R)患者的成纤维细胞中,线粒体耗氧量减少,呼吸链复合物活性降低[5,11,26,32,34-35,43]。ALS 相关突变R15L-CHCHD10 患者的成纤维细胞中,线粒体OXPHOS 水平与正常对照组相比显著降低。在突变患者的成纤维细胞中转染野生型CHCHD10,线粒体OXPHOS水平明显升高,可恢复到正常对照组的水平[32]。而且,CHCHD10 的ALS 相关疾病突变患者细胞中的线粒体受损情况,与在细胞中敲除CHCHD10 基因导致的结果一致[32]。然而,CHCHD10不同疾病突变体对线粒体OXPHOS的影响略有不同,如在G66V突变体患者的肌肉组织活检样本中,COX-negative的纤维不超过总纤维数的1%[27-28]。同样,G66V 突变患者的成纤维细胞中,呼吸链复合物活性及ATP 合成均无显著改变[36]。上述报道中均未提及CHCHD10具体以何种方式参与调节线粒体OXPHOS。
在8%低氧情况下,CHCHD10可进入细胞核,结合并活化转录抑制因子CXXC5(CXXC finger protein 5),进而下调在启动子处含有ORE 元件的基因表达,使COX4I2 蛋白表达下降[31]。在线粒体中,CHCHD10 可直接与COX6B 相互作用,也可作为支架蛋白,辅助ARG激酶调节CHCHD2磷酸化,进而活化COX[31]。而CHCHD10 的疾病相关突变体(如G66V、P80L)却失去支架作用,与CHCHD2的相互作用减弱,同时细胞耗氧量减少,ROS 水平增加[31]。此外,在细胞核中未能检测到CHCHD10 突变体(G66V、P80L)与CXXC5 相互作用,CHCHD10对ORE元件的抑制减弱,也会导致细胞耗氧量减少,ROS水平增加[31]。
5 CHCHD10引起神经退行性疾病的致病机理
5.1 CHCHD10单倍剂量不足
CHCHD10既可作为线粒体结构的组成部分参与线粒体嵴结构完整性的维持,也可作为调节因子调控线粒体呼吸链复合物蛋白质的表达水平。CHCHD10本身的表达降低会影响线粒体的结构和功能。在体外细胞实验中,敲减CHCHD10导致线粒体嵴结构发生异常[34,37],线粒体呼吸作用减弱,呼吸链复合物活性降低,ATP合成减少[10-11,34]。向CHCHD10 敲除的细胞中回补野生型CHCH10,线粒体OXPHOS 恢复至正常水平[32]。在TDP-43 转基因小鼠模型中,随着TDP-43 诱导神经退行性疾病的病程加重,皮层中CHCHD10蛋白的表达量随之降低[44]。在ALS 患者的组织样本中,也发现CHCHD10的mRNA水平和蛋白质表达量与对照组相比显著降低[44-45]。因此,CHCHD10单倍剂量不足或许与神经退行性疾病相关。
5.2 CHCHD10与CHCHD2均会发生突变
CHCHD10和CHCHD2均会发生突变,且与多种神经退行性疾病密切相关(图1),这些突变均会干扰二者之间的相互作用。CHCHD10 的突变(如G66V、P80L)在线粒体中不再作为支架蛋白,辅助ARG激酶调节CHCHD2的磷酸化,线粒体耗氧量减少,ROS 水平增加[31]。在细胞系中表达CHCHD2 的PD 相关突变体(如T61I、R145Q、Q126*),免疫共沉淀检测到CHCHD2 突变体与CHCHD10的相互作用明显降低[33]。但在不同细胞系中得到的实验结果存在差异,如在HEK293T 转染CHCHD2 突变体T61I,免疫共沉淀检测到突变体与CHCHD10 相互作用是增加的[46]。CHCHD10疾病突变体(如G58R、 S59L、 G66V) 和CHCHD2 疾病突 变 体T61I 都能促 进CHCHD2 与CHCHD10 形成异二聚体,导致蛋白聚集而损伤线粒体。因此,CHCHD2-CHCHD10异二聚体的形成可能是PD和/或ALS的新致病因素[34]。
5.3 MICOS复合物完整性缺失
MICOS复合物的核心组成部分Mic60/mitofilin与线粒体内膜蛋白OPA1相互作用,共同调节线粒体嵴结构的完整性。虽然CHCHD10 是否参与MICOS 复合物的组成还有待进一步确认,但CHCHD10 的缺失或突变确实会影响MICOS 复合物 的 完整 性。 在HEK293T 细 胞 中, 干 扰CHCHD10表达后,MICOS复合物的三个重要亚基mitofilin、Mic23 和Mic19 的蛋白质表达量与对照组相比显著降低。在细胞中敲减CHCHD10,使用非变性胶电泳(blue-native gel) 检测线粒体mitofilin-OPA1 二者形成复合物的多少,发现在720 ku 分子质量处mitofilin-OPA1 蛋白复合物减少了50%,另外在480 ku分子质量处mitofilin蛋白量同样减少了50%。在CHCHD10敲减细胞中,免疫共沉淀实验检测到mitofilin 与OPA1 之间的相互作用减弱,原位邻近连接技术(in situproximity ligation assay,PLA)检测到二者之间的相互作用减少了70%。在细胞系中转染野生型CHCHD10,与未转染的细胞相比,mitofilin 和OPA1 之间的结合增加接近2倍[41]。然而,CHCHD10疾病突变体(如R15L、S59L)会破环mitofilin 与OPA1 结合,mitofilin-OPA1复合物下降了 65%[41]。 在CHCHD10 转基因小鼠的海马中得到了同样的结果,CHCHD10 疾病突变体(如R15L、S59L)干扰mitofilin 与OPA1之间的结合[41]。 因 此,CHCHD10 缺失或突变破坏mitofilin-OPA1 复合物形成,影响MICOS 复合物的组装,最终破坏线粒体嵴结构的完整性。
5.4 OMA1过度激活
锌金属肽酶OMA1 是ATP 酶依赖的锌代谢酶,具有多个跨膜结构域,其锌指结构域位于线粒体内膜上[47]。OMA1 活化与线粒体膜电位的改变或线粒体应激有关[37]。OMA1 主要通过两种方式参与调节线粒体的形态结构。一种方式是OPA1 水解。在线粒体应激或者参与线粒体质量控制的蛋白酶(如YME1L、AFG3L2、SPG7)功能缺失的情况下,OMA1被激活[37],将L-OPA1水解成S-OPA1,L-OPA1/S-OPA1 比例失衡使线粒体分裂-融合发生改变,导致线粒体嵴的改变,表现为线粒体片段化[37]。在CHCHD2-CHCHD10 双基因敲除小鼠的原纤维细胞中,线粒体嵴呈异常的环形结构,而且,与对照小鼠相比,线粒体嵴的数目和面积均显著降低[37]。这种异常的线粒体结构与在MEF细胞中将OPA1敲除后的线粒体结构相似。在正常生理状态下,OPA1 被蛋白酶YME1 和OMA1 水解形成3条(c/d/e)S-OPA1蛋白条带。蛋白质免疫印迹实验证明,CHCHD2-CHCHD10双基因敲除小鼠的原纤维细胞中OMA1 蛋白酶被激活,OMA1 水解OPA1 形 成S-OPA1 (c/e)的比例增加[37]。在HEK293 细 胞 中, 将CHCHD2、 CHCHD10、OMA1三者同时敲除,线粒体嵴密度恢复至正常水平[37]。CHCHD10突变导致的线粒体片段化依赖于OMA1 水解OPA1 形成S-OPA1 的过程。比如,使用CHCHD10 突变体G58R 转染细胞,OMA1 即被激活,线粒体发生片段化。在转染G58RCHCHD10 的细胞中同时敲除OMA1,线粒体片段化程度降低[37]。因此,CHCHD2 和CHCHD10 缺失或突变导致线粒体嵴结构异常与OMA1 激活、OPA1 水解增加有关。OMA1 参与调节线粒体形态结构的另一种方式是线粒体整体应激反应(mitochondrial integrated stress response, mtISR)激活。 在G58R-CHCHD10 蛋白异常聚集或CHCHD10 基因单倍剂量不足时,线粒体应激使OMA1激活而切割DELE1 (DAP3-binding cell death enhancer 1),切割后的DELE1 释放到细胞质中与转录起始因子激酶HRI (eukaryotic translation initiation factor 2 alpha kinase 1)相互作用,使真核细胞转录起始因子2(eukaryotic initiation factor 2 alpha,eIF2α)磷酸化并进入细胞核,导致mtISR相关基因(包括ATF4、ATF5、CHOP)的mRNA上调,从而激活mtISR通路[48]。
5.5 线粒体能量代谢紊乱
CHCHD10 突变导致线粒体能量代谢发生障碍。在ALS 疾病相关突变S59L-CHCHD10 的转基因小鼠模型中,mtISR调节的mTORC1被激活[39],mtISR 涉及到的一碳(1C)代谢和色氨酸代谢升高,线粒体OXPHOS 发生障碍[39,49]。mtISR 激活与S59L-CHCHD10同CHCHD2形成异二聚体有关,CHCHD10突变体与CHCHD2形成的异二聚体激活OMA1,而OMA1 的活化使mtISR 激活。长期mtISR 激活诱导的代谢重排导致氨基乙磺酸耗竭,核苷酸代谢失衡,出现mtDNA 耗竭,最终引起心衰[49]。半乳糖处理R15L-CHCHD10突变患者的成纤维细胞,1C 代谢发生异常,然而mTORC1 下调[50]。CHCHD2-CHCHD10 双基因敲除也能激活mtISR,并导致心肌病[37]。CHCHD2-CHCHD10双基因敲除小鼠表现出的能量代谢功能缺失和CHCHD10的G58R或S59L转基因小鼠的毒性获得(gain of toxicity) 表 型, 均 与mtISR 密 切相关[37,39,48-49]。
5.6 PINK1-Parkin过度激活
PINK1通过招募Parkin到损伤的线粒体从而诱导线粒体自噬。正常情况下,PINK1进入线粒体膜间隙内会被线粒体PARL 蛋白(presenilinassociated rhomboid-like)切割,然后释放到细胞质中,最终被泛素蛋白酶体降解。当线粒体受损时,PINK1在线粒体外膜上积累并被磷酸化,激活的PINK1 招募Parkin[51]。随后Parkin 使线粒体外膜上的电压依赖的阴离子通道(voltage dependent anion channel 1,VDAC1)和线粒体融合蛋白1/2(mitofusin 1/2,Mfn1/2)发生泛素化,诱导线粒体自 噬[52]。 HeLa细胞中转染突变体S59LCHCHD10,线粒体的PINK1-Parkin 表达量与对照组相比显著升高,线粒体发生片段化。使用PINK1抑制剂处理细胞,抑制PINK1活性,可挽回S59LCHCHD10诱导的线粒体损伤。在S59L-CHCHD10突变患者成纤维细胞中抑制PINK1,可挽回S59LCHCHD10 突变导致的线粒体片段化[38]。在S59LCHCHD10 转基因果蝇模型中,线粒体长度减小,ATP 合成降低。在此模型中抑制PINK1 的表达后,线粒体的OXPHOS 增加,ATP 合成增加,提示S59L-CHCHD10 突变体可能是通过PINK1-Parkin自噬途径诱导细胞毒性;而抑制PINK1 活性可部分挽救S59L-CHCHD10 造成的线粒体损伤[38]。因此,在CHCHD10 突变体中,PINK1-Parkin 过度激活也会导致线粒体损伤及细胞毒性。
5.7 TDP-43在胞质和线粒体中异常积累
TDP-43 是RNA结合蛋白(RNA binding protein,RBP)。在正常情况下,TDP-43 蛋白定位于细胞核,参与多种RNA 代谢与加工过程,包括RNA 转录调控、RNA 前体剪接、RNA 稳定性维持,以及miRNA 等非编码RNA 的形成等[53]。在FTLD、ALS和部分AD患者的病理组织中,TDP-43在胞质中异常积累并形成包涵体,这是TDP-43 蛋白病的主要病理学特征[54-55]。在病理情况下,TDP-43 可从细胞核转移到细胞质,形成聚集体,并在某些因子的帮助下进入线粒体,导致线粒体嵴减少、膜电位降低、ROS 水平升高、ATP 产生下降,最终引起神经元死亡[56]。研究发现,CHCHD10 与TDP-43 发生相互作用。在TDP-43 转基因小鼠和ALS患者的病理样本中,CHCHD10表达量下降[41,44]。在细胞系中过表达TDP-43 后,CHCHD10进入细胞核[57]。在HeLa 细 胞 中,将CHCHD10 敲除或者转染CHCHD10 的突变体,TDP-43会在细胞质中聚集,并进入线粒体[57]。与对照组相比,TDP-43 与S59L-CHCHD10 突变体相互作用增强,CHCHD10 突变体导致TDP-43 蛋白形成不溶性的蛋白质聚集体,使其更多地进入线粒体,造成线粒体损伤[38]。根据曼德斯重叠系数(Mander's overlap coefficient,MOC)计算FTLD和AD 患者脑组织样本中磷酸化的TDP-43(p-TDP-43)与CHCHD10 的共定位系数,分别是0.75 和0.84,在FTLD 与AD 之间没有显著差异。相反,CHCHD10 与磷酸化的TDP-43 共定位系数为0.18,表明18%的CHCHD10 与p-TDP-43 存在共定位[58]。FTLD-TDP 患者脑组织样本经过RIPA缓冲液裂解后,检测可溶解和不可溶解的TDP-43、p-TDP-43 和CHCHD10 的含量,结果显示FTLDTDP样本中不溶性的TDP-43、 p-TDP-43 和CHCHD10 分别是非患病对照组的2.1 倍、2.4 倍和1.7 倍,皮尔森相关性系数(Pearson's correlation coefficient,PCC)分析显示,不溶性TDP-43 与不溶性的CHCHD10具有明显相关性(R=0.640 7)[58]。在TDP-43 转基因小鼠背景下,野生型CHCHD10使TDP-43磷酸化水平降低并抑制TDP-43聚集,减轻TDP-43 造成的病理学特征; 而突变型CHCHD10 可促进TDP-43 磷酸化,导致其聚集增加[38,58]。在ALS相关突变R15L-CHCHD10患者的脊髓解剖样本中,CHCHD10大量聚集,但未发现CHCHD10 聚集体与TDP-43 有明显共定位[59]。因此,TDP-43 错误定位并进入线粒体造成线粒体损伤可能也是CHCHD10相关疾病的致病因素。
6 展 望
CHCHD10的功能研究均从疾病角度出发,分析其突变体对线粒体结构和功能造成的影响,而对其正常生理功能以及CHCHD10蛋白本身性质的研究甚少。目前CHCHD10的生理功能主要表现在两方面。a. CHCHD10 是MICOS 复合物的组成部分,但是此观点仍然存在争议。首先,MICOS 复合物的具体组成部分至今仍未被完全鉴定。其次,所有结果均以蛋白质之间的相互作用作为实验手段,还没有其他方法证明CHCHD10 是否属于MICOS 复合物的组成部分。目前亟待发展新的实验方法进行解析,或者筛选出MICOS 复合物新的组成蛋白,进一步证明CHCHD10是否与MICOS 复合物有关。b. 在缺氧条件下,CHCHD10进入细胞核中,通过CXXC5 与ORE 结构域结合抑制COX4I2 的基因表达。使用不同细胞系进行实验,得到的实验结果也存在差异,说明选择合适的模型对于CHCHD10的功能研究尤为重要。CHCHD10是线粒体蛋白,虽然疾病相关突变对其进入线粒体影响不大,但是表达CHCHD10突变体的细胞,其线粒体嵴却表现出非常明显的异常。因此,研究CHCHD10蛋白的结构和功能,以及疾病相关突变体对其结构和功能的影响,不仅有助于揭示CHCHD10在维持线粒体嵴结构和稳定性中的重要作用,还将为理解CHCHD10 突变在相关神经退行性疾病中的致病机理提供重要线索。
