新生儿期Prader-Willi 综合征1 例报告
2023-05-11申明琪辛玥郑荣秀阚璇
申明琪,辛玥,郑荣秀,阚璇
(天津医科大学总医院儿科,天津 300052)
Prader-Willi 综合征(Prader-Willi syndrome,PWS)又称肌张力低下-智能障碍-性腺发育滞后-肥胖综合征、普拉德-威利综合征,由Prader 等[1]于1956 年首次报道,是最早被证实涉及基因组印记的遗传性疾病,为父源染色体15q11.2-q13 区域印记基因功能缺陷所致的一种多系统异常的复杂综合征,是导致人类致命性肥胖的常见疾病之一[2]。现将本院2021 年收治的1 例新生儿期PWS 病例报道如下。
1 病例资料
患儿,男,28 min,因“肤色发绀28 min 伴呼吸困难5 min”于2021 年6 月1 日收入天津医科大学总医院新生儿病房。患儿孕足月剖宫产,其母“高龄初产、妊娠合并子宫肌瘤”,因“胎儿窘迫?”行剖宫产娩出,羊水Ⅱ度,量约800 mL,胎盘、脐带无异常。患儿生后自主呼吸不规则,反应欠佳,肌张力稍低,全身肤色发绀,初步复苏后无好转,心率100 次/min,予吸氧、再次刺激后呼吸反应、肌张力、肤色均逐渐好转,但仍有口周、手足发绀,入院前5 min 患儿出现呼吸困难,收入新生儿科。
父母均39 岁,体健,否认家族遗传病史,母亲孕期规律产检,未发现异常。
入院时查体:体温36℃,呼吸55 次/min,心率126 次/min,血压66/42 mmHg(1 mmHg=0.133 kPa),体重3.63 kg,头围35 cm,身长52 cm。反应欠佳,呻吟、吐沫,口周、手足发绀,前囟1.5×1.5 cm2,平软,张力不高,鼻扇、三凹征阳性,双肺可闻及粗中湿鸣,心音有力,律齐,心前区未闻及明显杂音,腹软不胀,肠鸣音正常,四肢肌张力可,右手腕部下垂,双手可疑通贯掌,握持、拥抱反射可引出,吸吮、觅食反射弱引出。男婴外生殖器,阴茎长约2.3 cm,双侧阴囊发育欠佳,右侧睾丸未完全降至阴囊内,左侧睾丸未触及(图1)。手足暖,前臂毛细血管再充盈时间<3 s。
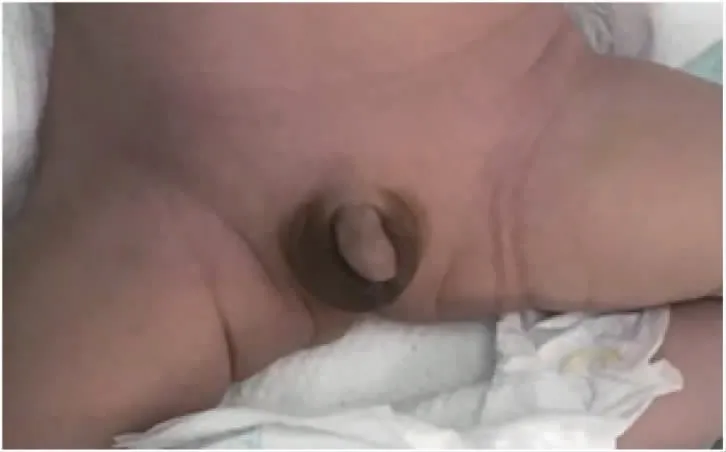
图1 左侧睾丸位置偏高,位于腹股沟内
辅助检查:血气分析电解质[双水平气道正压通气下:吸入氧浓度(FiO2)30%,呼气末正压(PEEP)5 cmH2O,吸气相正压(PIP)8 cmH2O]:pH 7.239,PO260 mmHg,PCO260.3 mmHg,Na+135.7 mmol/L,K+4.09 mmol/L,Ca2+1.392 mmol/L,Lac1.4 mmol/L,BE(ecf)-2.19 mmol/L,SO294.8%,Hct 50.1%。血常规:C 反应蛋白(CRP)、降钙素原(PCT)正常。肝肾功能、心肌酶:总蛋白47 g/L,白蛋白27 g/L,谷丙转氨酶35 U/L,谷草转氨酶78 U/L,碱性磷酸酶251 U/L,γ-谷氨酰转酞酶240 U/L,乳酸脱氢酶598 U/L,非结合胆红素32.8 μmol/L,结合胆红素0 μmol/L,尿素3.7 mmol/L,肌酐40 μmol/L,肌酸激酶711 U/L,肌酸激酶同工酶265 U/L。双手腕部X 线回报未见明显异常,不支持手腕畸形(图2)。胸片结果回报右肺透过度减低,多发片状模糊影,右肺纹理增多、模糊(图3)。颅脑超声回报未见明显异常。睾丸超声:左侧睾丸位置偏高,位于腹股沟内。腹部B 超未见明显异常。脐部超声:脐后下方低回声带(首先考虑为脐尿管残迹),外科会诊考虑脐尿管瘘。当日复查血气分析电解质正常。查血氨86.0 μmol/L。第10天查性激素全项:雌二醇<10 pg/mL,促卵泡生成素3.60 IU/L,孕酮0.59 ng/mL,促黄体生成素4.10 IU/L,睾酮122.99 ng/dL,催乳素160.09 ng/mL,符合小青春期水平。游离甲状腺功能未见异常。超声心动图示:卵圆孔开放状态。遗传、代谢病、血、尿筛査未见异常,外周血染色体回报:46,XY。病毒4 项无异常。4 h 振幅整合脑电图示脑电活动落后于矫正胎龄;24 h 动态脑电图结果回报:可见不连续型图形,低波幅段数秒。可见局部性低电压,右侧前额、额、前、中区显著。偶见爆发抑制样放电,异常脑电图。头颅核磁结果回报未见确切异常。第22 天肌电图:双下肢股神经潜伏期及左上肢尺神经运动传导速度大致正常;双上肢正中神经及双下肢股神经运动传导速度、腓总神经运动传导速度、动作电位波幅降低约20%~60%,传导速度轻度减慢;双下肢胫神经MCV 传导阻滞;双上肢正中神经感觉传导速度大致正常,双下肢胫后神经感觉传导速度明显减慢,显示多发性周围神经损害。出生后13 d 行基因检测:甲基化特异性多重连接探针扩增(MS-MLPA)方法显示15q11-13 区域基因拷贝数正常,但甲基化检测异常(图4),家系全外显基因检测显示为chr15母源单亲异二体,支持PWS 诊断(图5)。
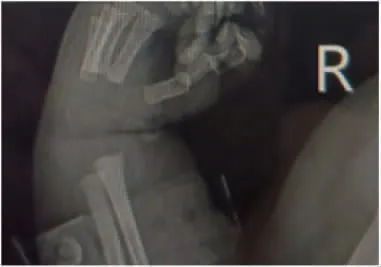
图2 右手腕X 片
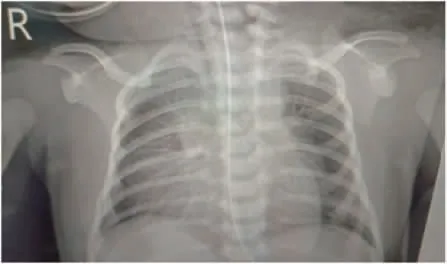
图3 入院胸片
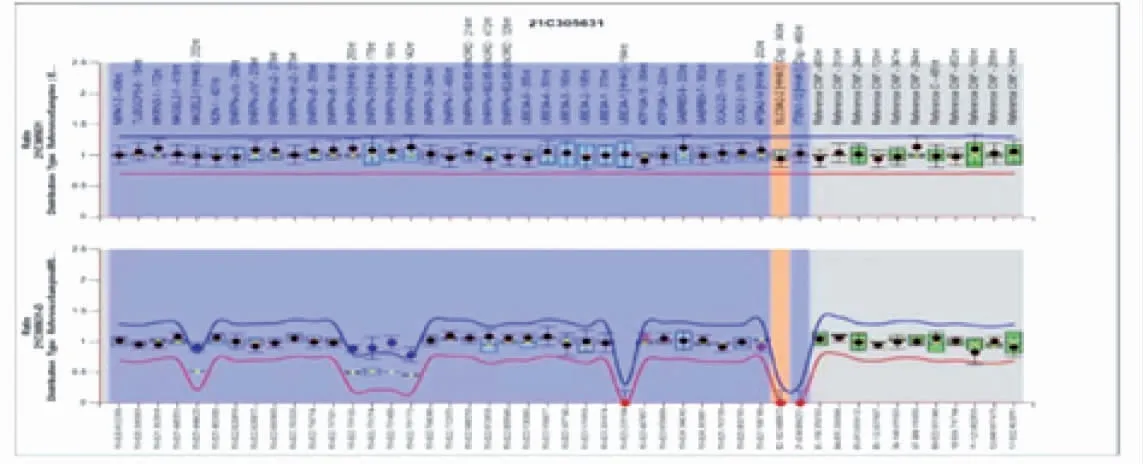
图4 MLPA 结果

图5 Trios 全外显子基因检测结果
治疗经过:入院后予无创呼吸机(BiPAP 模式)辅助通气2 d 后改为鼻导管吸氧3 d。予磷酸肌酸钠、心先安、维生素C 营养心肌治疗等。生后即反应欠佳、肌张力偏低、少哭、嗜睡,第7 天起反应及肌张力低逐渐好转,自主活动逐渐增多。第2 天起予配方奶喂养,因吸吮力弱配合鼻胃管喂养,第10 天起可自行吃奶全量,停用胃管。住院18 d,患儿生命体征平稳,吃奶慢,但可完全经口喂养,出院。
2 讨论
PWS 是一种多系统异常的复杂遗传病,估计患病率为1/20 000~1/10 000,报告范围为1/30 000~1/8 000。全球PWS 患者数量估计为400 000 例。大多数PWS 病例为散发病例,性别比约为1∶1。所有种族均有发病,但在白种人中报道更多。基于人群研究,PWS 中的死亡率大约为每年3%[2]。绝大多数PWS 为散发,其特点是智力残疾、行为问题、下丘脑功能障碍和特定畸形[3-4]。2015 年我国PWS 诊治专家共识提出了PWS 的临床诊断标准,同时指出PWS 临床表现因年龄而异[5]。胎儿期主要表现为胎动少;新生儿期主要表现为哭声弱、肌张力低下、吸吮无力、喂养困难,可致生长迟缓或停滞等。本例患儿生后哭声弱、肌张力低、喂养困难、吸吮无力需予管饲喂养,至生后18 d 可脱离管饲喂养。回顾分析93 例新生儿期诊断的PWS 患儿,其临床特点如下:男性59 例,女34 例;足月儿74 例,早产儿14 例,过期产儿2 例;剖宫产49 例,顺产17 例;宫内窘迫38 例,低出生体重儿15 例,皮肤色素缺失78 例,特殊面容40 例,男性隐睾44 例,女性小阴唇20 例;肌张力低下92 例,哭声微弱81 例,喂养困难65例,多采用MS-PCR 及MS-MLPA 方法检测,5 例患儿采用全基因芯片分析技术,7 例采用微阵列比较基因组杂交方法,检测结果为:73 例父源性15q11-q13染色体片段缺失,19 例母源性同源二倍体,1 例检测到SNORD116 基因缺失。这些患儿100%存在肌张力低下,88%存在哭声微弱,70%存在喂养困难。PWS 患儿婴幼儿期表现为食欲改善,喂养正常,生长速度恢复,体重增长,但可能出现生长发育及语言运动发育迟缓;儿童期表现为矮胖、小手小脚和认知功能障碍等;青春期表现为身高明显不足、肥胖、特殊性面容、智力残疾,学习困难、异常行为(包括强迫症、发脾气和自我伤害)以及生长和其他激素缺乏导致的性腺功能减退症[6]。肥胖及相关并发症是PWS 发病和死亡的主要原因,需要准确诊断、适当的医疗管理和治疗[2]。本例患儿生长发育各项指标里程碑明显落后:4 个月能够抬头,5 个月能够翻身,目前生后11 个月吃奶力度可,已添加辅食,身长82 cm,体重12 kg,但不能独坐,不会爬,不能独立站立。
分子遗传学研究发现,PWS 是一种神经发育基因组印记障碍,基因测序为诊断金标准。其主要有3种亚型,包括父系染色体15q11-q13 片段的缺失,是PWS 最常见的类型,约占65%~75%,或者母源15 号染色体单亲二倍体,约占20%~30%;或者印记中心微缺失或基因突变如SNORD116、SNURF、MAGEL2 等,占1%~3%[2-3]。我国PWS 患儿遗传类型73.5%为父源缺失型,26.5%为母源单亲二倍体[7]。本例患儿为15q11-13 区域基因拷贝数正常,但甲基化检测异常,家系全外显基因检测显示为chr15 母源单亲异二体。回顾分析的93 例新生儿期诊断的PWS 中父源染色体15q11-q13 片段的缺失占78%,稍高于既往报道,可能因为笔者设定的入选标准均为新生儿期诊断而致样本量偏少有关。
PWS 目前还没有治愈方法,可根据不同年龄段患儿的表型特征和不同的内分泌代谢紊乱及相关问题进行有效干预,需要多个学科的综合治疗。新生儿期超过80%的PWS 患儿出生后应注意给予胃管鼻饲喂养,以确保体重正常增加[8]。对于年长儿因食物强迫症而体重增加过多,标准治疗方法包括饮食限制、锻炼和生长激素替代[9]。不论是否合并矮小,建议在不存在明显生长激素使用禁忌证的情况下,宜早于2 岁开始生长激素治疗,以帮助肌肉组织发育、改善肌力和摄食能力[5]。接受生长激素治疗时年龄越小,智力及运动发育越好[10]。另外,应每天限制900 千卡的卡路里,锻炼计划侧重于日常有氧运动和姿势治疗[11]。
综上所述,PWS 发病率较低,单纯根据临床症状及常规化验检查易出现漏诊,需提高对本病的认识,并在早期完善分子遗传学检测,尽早明确诊断,早干预、早治疗,以期提高患儿的生存质量及生存率。对于新生儿时期即出现的不明原因的肌张力低下的患儿,如果同时出现了吸吮能力差、喂养困难、反应差、少动、哭声弱,均需要需高度警惕PWS的可能,尤其伴有性腺发育不良,特别是男性患儿伴有小阴囊、隐睾也是重要的临床线索,应及时完善基因检查明确诊断,以便早期干预,改善患儿的预后。在既往妊娠史曾明确有本病发生的夫妻,再次妊娠时应进行产前遗传咨询及诊断,以降低本病发生风险。
