Shwachman-Diamond综合征6例并文献复习
2023-05-07田凤艳李嘉董萧侯晓寒陈娇王叨魏琳琳张蕾刘玉峰
田凤艳,李嘉,董萧,侯晓寒,陈娇,王叨,魏琳琳,张蕾,刘玉峰
(郑州大学第一附属医院 儿科,河南 郑州 450052)
Shwachman-Diamond综合征(Shwachman-Diamond syndrome,SDS)是遗传性骨髓衰竭综合征的一种罕见亚型,发病率为1/153 000[1],以骨髓衰竭、胰腺外分泌功能不全和骨骼异常为主要临床特征。随着国内基因检测技术的发展,SDS诊断率逐渐提高,逐渐得到临床医生的重视,该病首发症状多样,极易漏诊、误诊,但国内文献报道总结病例数远低于其他国家,临床医生需加深对SDS的认识,提高诊断率。本研究回顾性分析SDS相关临床资料以加深对该病的认识。
1 资料与方法
1.1 资料收集收集2015年11月至2020年11月郑州大学第一附属医院收治的6例SDS患儿临床资料及院外持续随访情况。所有患儿均有SDS相关临床表现并经基因检测确诊为SDS[2]。本研究经医院医学伦理委员会审核批准(2022-KY-0946-002)。
1.2 分析方法收集6例SDS患儿临床资料,通过门诊复查和电话随访至2022年7月,了解患儿出院后的治疗及预后情况。对上述资料进行描述性分析,同时检索中国知网、万方数据库及PubMed数据库建库至2022年7月的相关文献,总结本病相关的基因突变、肿瘤易感机制及临床特点。
2 结果
2.1 临床表现6例患儿中5例男性,1例女性,于生后1~10 d发病,确诊年龄为1月余至13岁,6例患儿体格发育均明显落后,其中4例低于同年龄、同性别平均身高2个标准差(-2SD),符合矮小症的诊断标准[3],4例血细胞减少(1例粒缺+血小板减少,1例粒缺+轻度贫血,1例中度贫血,1例血小板减少),3例谷丙转氨酶升高,3例粪便可见脂肪滴,3例反复呼吸道感染,3例有腹泻/腹胀病史,2例有尿路感染病史。见表1。

表1 6例SDS患儿临床特征
2.2 实验室检查住院期间影像学检查提示,例2、3骨髓增生低下;例4、5骨髓增生活跃;例1四肢长骨X线示多发性骨密度异常(图1),既往外院CT示多发性长骨干骺端软骨发育不良;例2胸部CT示右肺上叶炎症,肺泡灌洗液镜检发现曲霉菌。入院时实验室检查结果见表2。

A为双侧股骨近端骨密度异常;B为左上肢尺骨桡骨近端、肱骨远端骨密度异常;C为右上肢尺骨桡骨近端、肱骨远端骨密度异常。
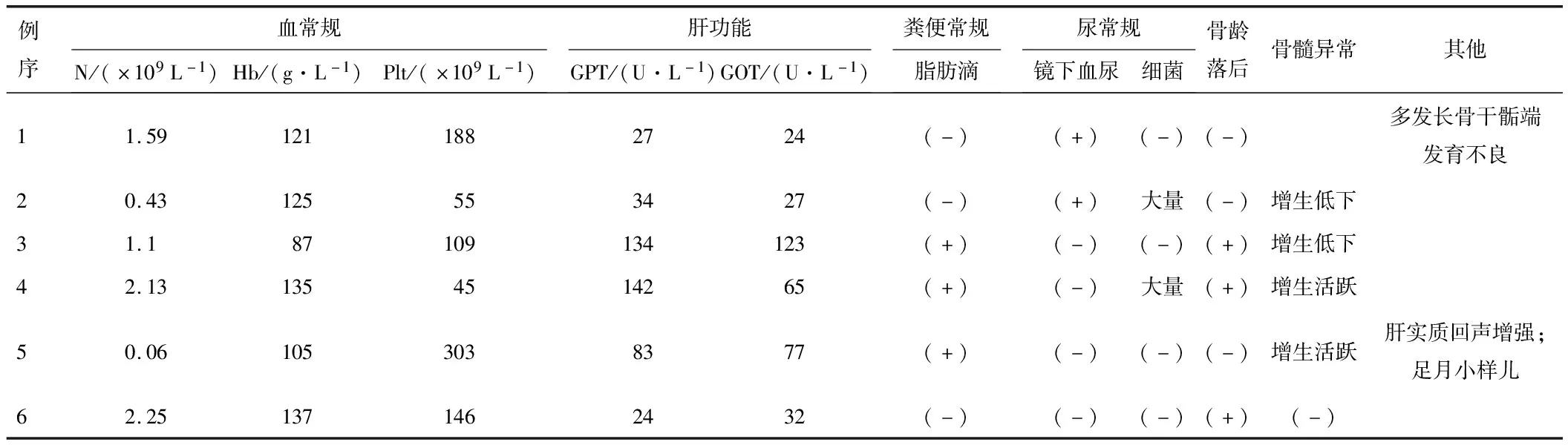
表2 6例SDS患儿主要辅助检查结果
2.3 治疗及随访情况例1~5均接受对症支持治疗,包括补充脂溶性维生素、钙剂、铁剂、胰酶制剂,口服熊去氧胆酸、保肝药,成分血输注,重组人粒细胞刺激因子、升血小板药物应用,口服激素,抗感染、抗病毒治疗。例6为例5先证后经家系验证确诊,给予胰酶制剂治疗腹泻。均监测血常规、肝功能、骨髓细胞形态。门诊复诊及电话随访至2022年7月,6例患儿肝功能均恢复正常,例1、2查血常规正常后未继续复查,例3、5、6复查血常规正常,例4于2018年12月行造血干细胞移植,2021年6月停用升血小板药物,复查血常规正常。例1~5随访身高均<-2SD,例3、5仍有脂肪泻、蛋白质吸收不良等胰腺外分泌功能不全表现,随访体重<3rd,例6随访身高为-1.2SD(13岁,150 cm)。
2.4 基因检测结果6例均为SBDS基因突变,例1SBDS基因为c.258+2T>C半合子突变,来自母亲,父亲SBDS基因exon1-2缺失突变;例2SBDS基因c.183_184delTAinsCT和c.258 +2T>C复合杂合突变,分别来自父母;例3SBDS基因c.258 +2T>C纯合突变,其父母均为c.258 +2T>C携带者;例4SBDS基因c.258+2T>C和c.184A>T复合杂合突变,分别来自父母;例5、6为亲兄弟,经家系基因检测为SBDS基因c.258+2T>C纯合突变,父亲、母亲均为携带者。见表3。

表3 6例SDS患儿基因测序情况
2.5 文献复习检索到国内外病例报道SDS患儿共546例(中文文献26篇,英文文献156篇)。国内包括本研究6例在内共59例SDS患儿,仅1例为SRP54基因突变,其余均为SBDS基因突变。我国SBDS基因突变常见核苷酸位点c.258+2T>C和c.183_184TA>CT复合杂合突变占32.7%(19/58),c.258+2T>C纯合突变占27.6%(16/58,其中7例发生恶性转化),c.258+2T>C和c.184A>T复合杂合突变占20.6%(12/58)。我国报道SDS确诊年龄中位数为1岁(生后1个月至16岁),33例记录明确就诊原因,其中 8例因反复感染就诊,17例因血细胞减少就诊,8例因肝功能异常就诊。我国SDS患儿中52例出现粒细胞减少,是国内SDS最常见的临床表现,28例出现腹泻(其中11例胰腺淀粉酶降低),26例身材矮小,22例存在骨骼异常,14例发生恶性转化,11例精神发育迟缓[4-14]。表4纳入国外资料完整的大型队列研究与我国所有SBDS突变病例以总结SBDS基因突变者分布特点[1,15-16]。

表4 各国SDS临床资料统计
此外,全球共30例SRP54突变,最常见的基因变异位点为c.349_351del(p.T117del)杂合突变,占50%(15/30)。93.3%(28/30)有血细胞减少,86.6%(26/30)骨髓涂片有早幼粒成熟停滞表现,86.6%(26/30)有反复感染病史,36.6%(11/30)神经发育及运动发育迟缓,20%(6/30)CT显示胰腺脂肪浸润,1例已发生恶性转化[17-18]。19例DNAJC21基因突变均有全血细胞减少,伴生长受限17例,运动发育迟缓8例,面部畸形8例,复发性感染8例,牙齿异常9例,胰腺功能障碍6例,视网膜营养不良6例,先天性髋关节发育不良6例,隐睾3例,短端粒7例[19-20]。10例EFL1基因突变,均有血细胞减少、神经发育迟缓、慢性腹泻,5例影像学显示胰腺脂肪浸润(2例胰酶替代后好转),8例干骺端软骨发育不良,5例矮小,5例肝功能异常(其中2例肝病理活检示肝硬化),3例死亡,死亡中位年龄为14.5月龄[21]。
3 讨论
Shwachman-Diamond综合征是由SBDS、SRP54、DNAJC21、EFL1这4种基因突变引起的多系统性罕见病[18,20-22],其中SRP54基因突变为常染色体显性遗传,其余3种基因突变为常染色体隐性遗传[23]。上述4种基因所编码的同名蛋白均参与核糖体成熟:细胞质中游离60S核糖体亚基前体与EIF6结合,处于无法翻译的非活性状态,核糖体成熟因子SBDS与EFL1协同促进EIF6从60S核糖体亚基前体解离,游离的60S核糖体亚基前体穿梭至细胞核成熟并与40S核糖体亚基结合形成80S核糖体,DNAJC21则起到稳定80S核糖体的作用,80S核糖体激活核糖体进行翻译,SRP54促进翻译所合成多肽的运输,这4种基因发生突变时,核糖体成熟被破坏,故SDS的发病主要与核糖体成熟的破坏有关[24]。文献复习发现男性发病数量明显高于女性,与本研究以男性为主相符。本研究6例均为SBDS基因突变,占比最高的基因突变为c.258+2T>C突变,与各国大型队列研究结果一致,SBDS基因突变导致同名蛋白SBDS耗竭无法解离MDM2-p53复合物,发挥肿瘤抑制作用,增加肿瘤易感性[25]。同时,40%的骨髓增生异常综合征存在SBDS突变,常伴有可驱动增加白血病发病可能途径的TP53突变,存在该突变的病例发生髓系恶性转化的风险更高且预后比其他病因所致的恶性髓系血液病更差[26]。
SDS以骨髓衰竭、胰腺外分泌功能障碍、骨骼异常为特征,临床可同时伴有消化、神经、内分泌等多系统受累。骨髓衰竭可表现为单系或多系血细胞减少、骨髓细胞学异常。本研究例2为中性粒细胞缺乏和血小板减少,例3以血红蛋白中度减少为主,例4以血小板减少为主,例5为中性粒细胞和血红蛋白减少,均于婴幼儿期出现,与文献复习结果一致。骨髓细胞学检查示骨髓造血能力下降或病态造血。例4已行造血干细胞移植,随访血常规三系正常。以血细胞减少为起病表现的SDS患儿极易出现严重的血液系统并发症,应监测血常规、骨髓造血情况,应尽早行造血干细胞移植,其远期预后需更多循证医学证据[15]。
脂肪泻/不耐受脂肪食物在婴幼儿时期最显著,同时生长发育明显受限,为胰腺外分泌功能障碍典型表现[1],患儿常首诊于儿童消化科,粪便常规检查示粪便脂肪滴阳性,影像学检查示胰腺脂肪化,血胰腺淀粉酶水平降低可协诊,但易漏诊SDS。婴幼儿可更换为深度水解奶粉减轻腹泻症状,胰腺外分泌功能障碍常导致脂溶性维生素缺乏甚至凝血功能异常、骨质疏松、慢性肝病等,应及时补充胰酶、益生菌、脂溶性维生素、钙剂等,但要警惕长期过量补充脂溶性维生素导致肝纤维化、泌尿系结石等。本研究例3、4、5确诊时粪便有脂肪滴,均有腹泻/腹胀表现,例1、2、6因婴幼儿时期未能诊断腹泻病因,SDS确诊年龄明显晚于其他3名患儿。通过随访,本研究例1、2、4、6腹泻症状均随年龄增长缓解,例3、5给予治疗后目前仍有间断腹泻,与文献中部分患儿学龄前期自发出现胰腺外分泌功能障碍的明显改善相一致[27-28]。本研究6例患儿均有体格发育的显著落后,且落后程度与胰腺外分泌功能不全程度相关,与大型队列研究显示的SDS患儿矮小症的易感性相符[29]。国外1例SDS患儿在应用重组人生长激素治疗2 a后,年生长速度和身高标准差评分均显著改善,但由于重组人生长激素有诱发肿瘤可能,国内尚未使用该方法治疗SDS病例[30]。
SDS的骨骼病变表现多样,包括四肢/胸廓/肋骨异常、脊柱侧弯、牙釉质发育不良、干骺端发育异常等,骨骼病变导致肢体畸形时需通过外科手术矫正[31]。本研究例1有四肢多发性骨密度异常及既往外院CT示干骺端软骨发育不良,予钙剂及维生素D口服治疗。此外,还有部分SDS病例可因肝功能异常起病首诊于儿童消化科,多数患儿肝功能异常可随年龄增长而恢复正常,但亦有部分病例发展成肝硬化、肝衰竭[32]。本研究例3、4、5曾有肝功能异常,定期检测给予保肝药治疗后恢复正常,确诊年龄较大的例1、例2肝功能正常。本研究例1、例5有专注力及语言理解能力落后等神经发育迟缓表现,与国内外SDS病例存在神经发育迟缓表现的结果相符。Perobelli等[33]发现SDS患儿的认知障碍与灰质和白质连接的弥漫性脑异常有关:与健康同年龄同性别儿童相比,SDS患儿头颅MRI显示大脑皮质厚度增大,左缘-前扣带回皮质厚度增加≥43%,但大脑左半球Broca’s区皮质明显变薄,且在语言和智力表现测试中评分显著降低。
总之,单一或同时出现血细胞减少、脂肪泻、生长发育受限、骨骼异常、肝功能异常伴或不伴反复感染的患儿就诊时,都应警惕SDS的可能,在出现典型表现前及早进行遗传学检查确诊,当未检测到SBDS基因突变应完善其余SDS相关基因检测,该病常呈家系分布,有先证者时建议进一步行家系基因检测以避免漏诊。确诊后除给予对症及支持治疗,必须定期监测血液系统,警惕恶性转化可能,早期诊断并于恶性转化前及早行造血干细胞移植。
