β-内酰胺酶抑制剂复方制剂非临床研究技术指南
2023-04-29卞星晨黄志伟胡付品武晓捷李鑫张菁
卞星晨 黄志伟 胡付品 武晓捷 李鑫 张菁
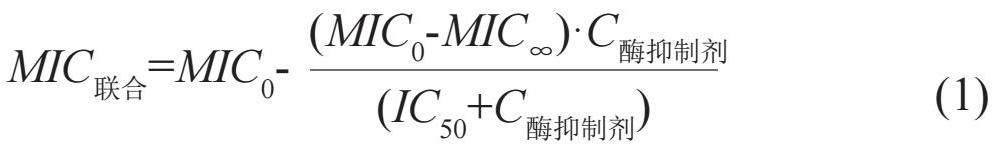


摘要:β-內酰胺酶抑制剂复方制剂在临床上被广泛应用于治疗耐药菌所致感染,由于早期β-内酰胺酶抑制剂的抑酶谱较窄,抑酶谱更广泛的酶抑制剂在不断研发之中。与一般抗菌药物临床前研究不同,β-内酰胺酶抑制剂复方制剂的临床前研究需明确β-内酰胺类药物或酶抑制剂本身的抗菌谱与抗菌活性,尤其是明确酶抑制剂是否具有抗菌活性。需要确定合适的β-内酰胺类药物与酶抑制剂复方制剂,以及适用的不同酶型的目标病原菌。本文主要介绍新型β-内酰胺酶抑制剂复方制剂临床前研究方法。临床前研究阶段的β-内酰胺酶抑制剂复方制剂研究包括体外研究和体内研究两部分,前者主要为体外药效学研究和体外药动学/药效学(pharmacokinetic/pharmacodynamic, PK/PD)研究,常用研究方法包括β-内酰胺类药物和β-内酰胺酶抑制剂复方制剂最低抑菌浓度测定、最低杀菌浓度测定、抗生素后效应测定及时间杀菌曲线。后者主要为动物药动学研究、感染动物药效学研究和感染动物药动学/药效学研究。在动物药动学/药效学研究中,需考虑β-内酰胺类药物与酶抑制剂的相互影响。这些研究方法的应用旨在阐明β-内酰胺酶抑制剂复方制剂两组分药效学特点、药动学相似与否、PK/PD指数及其临床前PK/PD靶值,为进入临床试验阶段目标适应症及剂量选择提供依据。
关键词:β-内酰胺酶抑制剂复方制剂;药动学;药效学;药动学/药效学靶值
中图分类号:R978.1文献标志码:A
Abstract β-lactam/β-lactamase inhibitor combinations are widely used in clinical treatment of resistant bacterial infections. The early-developed β-lactamase inhibitors have narrow inhibitory spectrum, those with broader inhibitory spectrums are being developed. Different from general antimicrobials, preclinical studies of β-lactam/β-lactamase inhibitor combinations need to clarify the antimicrobial spectrum and activities of β-lactam or β-lactamase inhibitors, especially whether β-lactamase inhibitors have antimicrobial activities. Its necessary to determine the appropriate β- lactam and β-lactamase inhibitor combinations, as well as the applicable range of target pathogens with different β-lactam types. The technical guidelines mainly introduce preclinical research methods of new β-lactam/β-lactamase inhibitor combinations. Preclinical studies of β-lactam/β-lactamase inhibitor combinations include in vitro and in vivo sections. The former mainly refers to in vitro pharmacodynamics(PD)and pharmacokinetic/pharmacodynamic(PK/PD)studies. Common methods include minimum inhibitory concentration and minimum bactericidal concentration determinations, post-antibiotic effect determination and time-killing curve assay of β-lactam/β-lactamase inhibitor combinations. The latter mainly refers to PK studies in animals, PD and PK/PD studies in infected animals. In animal PK/PD studies, the interaction between β-lactams and β-lactamase inhibitors should be considered. The purpose is to elucidate the pharmacodynamic properties, pharmacokinetic similarities of β-lactam and β-lactamase inhibitor in the studied combination, and to determine the PK/PD indices and preclinical PK/PD target values, and hence to provide basis for target indications and dose selection in clinical trial.
Key words β-lactam/β-lactamase inhibitor combinations;Pharmacokinetics;Pharmacodynamics;Pharmacokinetic/pharmacodynamic target values
各种β-内酰胺酶的产生是绝大部分革兰阴性菌对β-内酰胺类抗生素耐药的最重要机制,主要可分为丝氨酸酶(A类、C类和D类酶)和金属酶(B类酶)两大类。为了避免β-内酰胺类抗生素尤其是碳青霉烯类药物被水解而失活,β-内酰胺类抗生素/β-内酰胺酶抑制剂复方制剂(简称β-内酰胺酶抑制剂复方制剂)应运而生,目的在于恢复现有抗菌药物活性,保护有效的治疗药物,延长使用周期[1]。早期研发的酶抑制剂克拉维酸、他唑巴坦与舒巴坦仅能覆盖A类酶,近年来国外已上市了头孢洛扎-他唑巴坦(2:1)、头孢他啶-阿维巴坦(4:1)、美罗培南-法硼巴坦(1:1)及亚胺培南-西司他丁/雷利巴坦(2:2:1)4种酶抑制剂复方制剂,覆盖酶类扩展到C类与D类酶[1]。我国也在加速研发新型β-内酰胺酶抑制剂复方制剂。
抗菌药物研发最大的特点是可以通过临床前研究指导临床研究。相比于单药抗菌药物,β-内酰胺酶抑制剂复方制剂的临床前研究内容与方法均有所差异,包括明确β-内酰胺类药物或酶抑制剂本身抗菌谱与抗菌活性,确定合适的β-内酰胺类药物与酶抑制剂组合,以及适用的目标酶型病原菌,进行动物体内两药药动学(pharmacokinetics, PK)研究,评价PK相似性,在PK/PD研究中需考虑β-内酰胺类药物和酶抑制剂间的相互影响、两药联合最佳配比等。为此,本文在国家药品审评中心2017年颁布的《抗菌药物药动学/药效学研究技术指导原则》基础上,结合已上市酶抑制剂复方制剂的临床前研发公布数据及相关文献,着重介绍β-内酰胺酶抑制剂复方制剂与一般抗菌药物临床前研究的差异性与关注点,以期为我国新型β-内酰胺酶抑制剂复方制剂的临床前研发提供参考。
1 概述
1.1 β-内酰胺酶抑制剂复方制剂临床前研究內容
β-内酰胺酶抑制剂复方制剂的临床前研究包括体外研究和体内研究两部分,前者主要为体外PD研究和体外PK/PD研究,常用研究方法包括最低抑菌浓度(minimum inhibitory concentration, MIC)测定、最低杀菌浓度(minimum bactericidal concentration, MBC)测定、抗生素后效应(post-antibiotic effect, PAE)测定及时间杀菌曲线。后者主要为动物PK研究、感染动物PD研究和感染动物PK/PD研究[1]。
1.2 临床前研究目的、应用范围
β-内酰胺酶抑制剂复方制剂的临床前研究目的包括以下几个方面:明确β-内酰胺类药物及酶抑制剂单独应用与作为复方制剂联用时的抗菌谱、抗菌活性与最佳配比。揭示β-内酰胺类药物及酶抑制剂在动物体内的PK特征,评价两药联合对感染动物的疗效,获得两药PK/PD指数与靶值。
2体外研究
2.1 体外药效学研究
本部分研究旨在明确β-内酰胺类药物及酶抑制剂单独应用与作为复方制剂联用时的抗菌谱、抗菌活性等药效学特征,主要评价参数、技术方法和要求及结果描述如下。
2.1.1 单药抗菌活性
受试菌的选择、测定的总株数、受试浓度及测定方法参见《抗菌药物药效学非临床研究技术指南》[2]。
2.1.2 联合抗菌活性
采用棋盘法开展β-内酰胺类药物与酶抑制剂联合药敏试验。酶抑制剂在大多数情况下无抗菌活性,无法采用部分抑菌浓度指数(fractional inhibitory concentration index, FICI)评价协同作用。可引入模型法,设计一系列β-内酰胺类药物与酶抑制剂的浓度梯度,获得两药协同抑菌浓度后,采用inhibitory Emax模型(公式1),建立联合方案下β-内酰胺类药物MIC值与酶抑制剂浓度的关系[3]。结果描述包括无酶抑制剂存在下和酶抑制剂浓度趋向无穷大时β-内酰胺类药物最低抑菌浓度MIC0和MIC∞,反映酶抑制剂抑酶活性的浓度比值MIC0/MIC∞,50%和90%最大抑菌作用下酶抑制剂的浓度IC50和IC90,β-内酰胺类药物MIC值-酶抑制剂浓度拟合效果图等。
2.1.3 酶抑制剂对β-内酰胺酶的体外抑制活性
选取常见β-内酰胺酶类型,测定酶抑制剂对四类β-内酰胺酶的体外抑制活性,包括丝氨酸酶(A类、C类和D类酶)及金属酶(B类酶)。酶种类应具有较高的临床检出率与代表性,结果描述参数为IC50值,即抑制50% β-内酰胺酶所需酶抑制剂浓度,确定该酶抑制剂针对哪一种产酶菌株具有最佳抑制效果。
2.1.4 β-内酰胺酶抑制剂复方制剂最佳配比
β-内酰胺酶抑制剂复方制剂配比筛选一般采用棋盘法联合药敏试验,两药以不同系列的浓度进行混合,过夜孵育后阅读结果,获得两药最佳配比,包括固定比例配比和固定浓度配比。固定比例配比一般可涵盖8:1、4:1、2:1、1:1[4],固定浓度时酶抑制剂浓度范围一般为2~8 mg/L。
除体外药效学研究,体外PK/PD模型及感染动物PD研究也可用于以上两药最佳配比的筛选,以菌落数下降作为药效学指标。
2.1.5 β-内酰胺类药物最低杀菌浓度
受试菌的选择、测定的总株数及测定方法参见《抗菌药物药效学非临床研究技术指南》[2]。
2.1.6 β-内酰胺类药物抗生素后效应
受试菌的选择、测定方法参见《抗菌药物药效学非临床研究技术指南》[2]。
2.1.7 时间杀菌曲线
时间杀菌曲线(time-killing curve)通常指静态杀菌曲线,是指在β-内酰胺类药物及酶抑制剂单药及联合应用下,选择合适的两药配比,观察药物对受试菌的杀菌活性及杀菌速率随浓度的变化过程。联合方案中的药物配比可参照“2.1.4”筛选获得的最佳配比或固定酶抑制剂浓度。受试菌选择、结果描述参见《抗菌药物药效学非临床研究技术指南》[2]。
2.2 体外PK/PD模型
抗菌药物体外PK/PD模型(in vitro PK/PD model)借助体外装置模拟抗菌药物在机体内药物浓度随时间变化(药动学过程),在动态药物浓度下研究抗菌药物对细菌的抑制或杀灭作用(药效学)。此模型可用于抗菌药物体外PK/PD指数及靶值的制定、给药方案的筛选,尤其适用于基于细菌耐药机制的抗菌药物PK/PD研究。研究持续时间取决于研究目的,对于药物的PK/PD指数相关研究,给药持续时间通常为1~3 d,以24 h最为多见。如研究不同给药方案下细菌耐药性的发生发展,给药持续时间应模拟临床上该适应证的疗程,通常至少持续5~8 d。体外研究的实施有助于减少后续实验动物的消耗,减小临床试验的规模[5]。有关于模型分类,目标病原菌的选择及药动学模拟参考《抗菌药物药动学/药效学研究技术指导原则》[6]。
2.3 基于机制的PK/PD模型
获得体外静态或动态杀菌实验数据后,可建立基于机制的PK/PD模型,用于预测不同给药方案对目标病原菌的杀菌效果[7]。考虑到敏感菌和耐药菌对药物的反应不同,细菌通常被分为敏感和耐药两个亚群,模型构成包括细菌生长、β-内酰胺类药物自身的杀菌作用,β-内酰胺酶对药物的降解,酶抑制剂对其杀菌作用的增强,对β-内酰胺酶的抑制作用,如酶抑制剂本身有抗菌活性也需要考虑在内。
3 体内研究
动物感染模型(in vivo PK/PD model)可用于研究β-内酰胺酶抑制剂复方制剂体内PK特点、抑菌或杀菌作用,据此获得的动物PK/PD指数及靶值可外推至人体,其结果与临床研究结果有较好的一致性。现有动物感染模型如大腿感染、尿路感染、肺炎、腹腔感染、心内膜炎和全身感染模型等,已用于目标适应证为细菌性肺炎、尿路感染、血液感染、复杂性腹腔感染和皮肤软组织感染等抗菌新药研发中[6]。
选择的动物PK/PD模型应与β-内酰胺酶抑制剂复方制剂可能治疗的感染类型有很好的一致性。模型建立时,采用的感染动物一般为小鼠、大鼠,对于一些较复杂的模型如脑膜炎、心内膜炎等可采用兔,且通常同时在免疫抑制感染动物和免疫正常感染动物体内评价β-内酰胺酶抑制剂复方制剂体内活性。通过腹腔注射环磷酰胺的方法进行免疫抑制,常用的免疫抑制鼠大腿感染模型和肺炎模型,其药效判断指标明确(组织菌落计数对数值变化)、重复性好且操作简单。药动学参数一般来源于血浆/血清药物浓度,可采用微透析技术测定靶部位的药物浓度动态变化,比较和评价药物在血液及靶组织中的PK/PD特性[6]。
感染菌株的选择一般需包括标准菌株和临床菌株、敏感菌株和耐药菌株。对于耐药菌株需采用分子生物学方法确定菌株携带的耐药基因及产β-内酰胺酶类型,明確其遗传背景。
获得感染动物模型中PK/PD靶值的关键步骤包括β-内酰胺酶抑制剂复方制剂给药方案的设计、药物体内暴露量的准确测定、药效学指标的选择和体内PK/PD指数的选择等。
3.1 动物PK研究
动物PK研究内容、要求及结果分析等详见NMPA颁布的《药物临床前药动学研究技术指导原则》[8]。一般在感染动物模型中测定β-内酰胺类药物及酶抑制剂在动物体内不同时间点血浆(血清)浓度,计算两药单药及联合后不同剂量和配比、不同给药频率给药后动物的PK参数,比较β-内酰胺类药物及酶抑制剂的消除半衰期是否接近,较接近者适合作为复方制剂联合应用。实验组需设立单独给药组与联合给药组,考察两药之间的PK是否存在相互作用,联合时的PK是否发生改变。当感染状态影响药物在组织中的穿透性时,需研究感染动物与正常动物PK的差异。对于使用环磷酰胺进行免疫抑制的动物,应说明其对于动物PK的可能影响。对于某些动物组织或部位,如中枢神经系统、肺和皮肤,其药物浓度可能明显不同于血浆/血清中的浓度,因此,除血浆/血清中药物浓度外,建议提供该感染部位中药物浓度数据和组织体液穿透率等。
3.2 感染动物PD研究
本部分研究旨在筛选可获得最佳杀菌效果的β-内酰胺类药物与酶抑制剂的剂量与配比。通常采用小鼠大腿或肺部感染模型,选取β-内酰胺类药物主要目标适应证覆盖的目标细菌,若酶抑制剂为新药,依据体外筛选结果选取主要抑制的产酶类型菌株,需包含不同MIC值的细菌。实验组的设计分以下几种情况:①β-内酰胺类药物为新药,设立治疗组(β-内酰胺类药物单药及复方制剂)与对照组(未给予药物治疗者);②酶抑制剂为新药且本身无抗菌活性,实验组同①,本身有抗菌活性实验组在①基础上增加一组酶抑制剂单药;③两药均为新药,治疗组应包括两药单药以及作为复方制剂应用,另设立对照组。通常先进行预实验摸索给药剂量,给药剂量范围需涵盖无效至最大药效剂量,两药配比可涵盖8:1、4:1、2:1、1:1,以不同频率给药,给药后不同时间进行感染动物局部组织或血液中细菌菌落计数,记录感染动物存活率/死亡率及存活天数等药效学指标,确定最佳用药剂量及配比。
确定最佳给药方案后,选取更多不同种属细菌,比较β-内酰胺类药物单药及联合酶抑制剂对各种属目标细菌的杀菌效果,据此进行药效学评价。
3.3 感染动物PK/PD研究
该研究通过建立数学模型定量描述抗菌药物不同给药方案下感染动物体内药物暴露量随时间变化以及与药效学指标之间的关系,药效学指标包括动物体内细菌菌落计数变化、动物存活率或死亡率,菌落计数变化由于定量方便准确更常被采用。一般需要3~5株研究菌株,覆盖主要的目标适应证细菌,需选取MIC较高的野生型菌株,应覆盖MIC50和MIC90的菌株。感染动物模型可用于评价抗菌药物不同给药方案(剂量、间隔、治疗天数)下取得的微生物学疗效(感染血液/组织中细菌菌落数降低)和治疗效果(动物存活率),剂量组设计应涵盖尽可能宽的范围,更有利于Emax模型的准确拟合,据此推荐预期人体内最大杀菌效果和临床疗效的给药方案,为后续临床试验有效剂量范围的选择提供参考。
4 PK/PD靶值研究
4.1 β-内酰胺类药物单独应用的PK/PD靶值
设计β-内酰胺类药物剂量递增和剂量分割实验,结合该药PK参数如Cmax、AUC0-24,蛋白结合率及受试菌MIC值,建立3个PK/PD指数fCmax/MIC、fAUC0-24/MIC和f%T>MIC与其药效学参数(细菌菌落计数变化值,ΔLog10CFU)的药效学模型(如Sigmoidal Emax模型,公式2)。根据相关性大小(R2)选择代表该β-内酰胺类药物的最佳体外PK/PD指数,多数β-内酰胺类药物最佳体外PK/PD指数为f%T>MIC。将细菌菌落计数对数降低不同单位时(ΔLog10CFU取值0、-1或-2,分别对应细菌净生长为零、细菌菌落计数降低至1/10和1/100)fCmax/MIC、fAUC0-24/MIC或f%T>MIC的对应值作为PK/PD靶值。可合并同种菌株计算PK/PD靶值,或逐个菌株进行分析,并对PK/PD靶值作描述性统计分析。结果描述中需包括细菌菌落计数变化值-PK/PD指数图(图1)。
其中E0为细菌基线计数值,Emax为最大杀菌效力、EC50为达最大杀菌效力一半时的PK/PD指数值、γ为希尔系数(反映曲线陡度),X为PK/PD指数值。
4.2 β-内酰胺类药物与酶抑制剂联合的PK/PD靶值
为明确β-内酰胺类药物与酶抑制剂联合时其PK/PD靶值是否发生改变,可开展本部分研究。一般酶抑制剂维持恒定浓度或固定剂量,β-内酰胺类药物依据单独应用时靶值制定流程[9]。
4.3 β-内酰胺酶抑制剂的PK/PD靶值
如酶抑制剂本身对细菌具有抗菌活性(如舒巴坦对鲍曼不动杆菌有一定抗菌活性),则参照β-内酰胺类药物PK/PD靶值制定流程。考虑到大部分酶抑制剂本身不具有抗菌活性,一般需联合β-内酰胺类药物。β-内酰胺类药物以合适的固定剂量给药,与空白对照组细菌生长相当的最高剂量为佳,β-内酰胺类药物剂量过大可能掩盖酶抑制剂的剂量效应关系[10-11]。
对酶抑制剂进行剂量递增和剂量分割实验,流程参考4.1部分。由于酶抑制剂抗菌活性的多样性,其PK/PD指数有多种可能,阿维巴坦和他唑巴坦PK/PD指数为f%T>CT,法硼巴坦和雷利巴坦PK/PD指数为fAUC0-24/MIC(此处MIC指联合8 mg/L法硼巴坦时美罗培南MIC或联合4 mg/L雷利巴坦时亚胺培南MIC,如有酶抑制剂人体PK研究可参考稳态坪浓度确定其在MIC测定中的固定浓度)[11-12]。CT值为可抑制β-内酰胺酶活性,保证β-内酰胺类药物不被水解的酶抑制剂浓度,也可为與药效学参数最相关的浓度值(即相关性大小R2最高)[13]。CT值的确定:建立中空纤维感染模型,β-内酰胺类药物以恒定浓度恒速加入,改变酶抑制剂的浓度,依据受试细菌发生再生长时间点时的酶抑制剂浓度估计CT值的范围[13];或选取数个CT值,对每一个CT值进行模型拟合,以拟合效果最佳的浓度作为CT值[14]。
4.4 注意事项
β-内酰胺酶抑制剂复方制剂PK/PD研究应注意选择合适范围的剂量组。剂量选择不宜过窄,窄剂量如果落在PK/PD指数低处,可能导致无法精确估计Emax(图1-A),以至于远远高估Emax;窄剂量如果落在EC50处,可能导致Emax模型无法拟合(图1-B);窄剂量如果落在PK/PD指数高处,可能导致无法精确估计EC50和希尔系数γ(图1-C),导致EC50估计远远低于真实值。理想的Emax模型拟合图应能精确估计各个参数(图1-D)。因此,除了考虑PK/PD指数与抗菌效应的相关性,也应注重模型参数估计的准确性。
在探索β-内酰胺酶抑制剂复方制剂的PK/PD指数时,应探索多种PK/PD指数的可能性,包括但不限于fAUC、fCmax、f%T>CT(f %T>0.25 mg/L、%T>1 mg/L、%T>4 mg/L和%T>8 mg/L等)、fAUC/MIC、fCmax/MIC等,寻找与抗菌效应最相关的PK/PD指数[11,15]。此外,体外PK/PD模型所估计的PK/PD靶值可能与动物研究或者临床研究的PK/PD靶值不一致,三者之间不能完全等价。
参 考 文 献
俞云松等. β-内酰胺类抗生素/β-内酰胺酶抑制剂复方制剂临床应用专家共识[J]. 中华医学杂志, 2020, 100(10): 738-747.
李聪然,杨信怡,王秀坤等. 抗菌药物药效学非临床研究技术指南[J]. 中国抗生素杂志, 2023, 48(1): 1-7.
Chauzy A, Buyck J, De Jonge B L M, et al. Pharmacodynamic modelling of beta-lactam/beta-lactamase inhibitor checkerboard data: illustration with aztreonam-avibactam[J]. Clin Microbiol Infect, 2019, 25(4): 515 e1- e4.
Livermore D M, Mushtaq S. Activity of biapenem (RPX2003) combined with the boronate beta-lactamase inhibitor RPX7009 against carbapenem-resistant Enterobacteriaceae[J]. J Antimicrob Chemother, 2013, 68(8): 1825-1831.
张菁. 药动学-药效学: 理论与应用[M]. 科学出版社, 2021.
NMPA. 抗菌药物药代动力学/药效学研究技术指导原则 [EB/OL]. https://www.cde.org.cn/zdyz/domesticinfopage?zdyzIdCODE=000fb29bc72a100e6f266433e4d7be5c.
Chauzy A, Gaelzer Silva Torres B, Buyck J, et al. Semimechanistic pharmacodynamic modeling of aztreonam-avibactam combination to understand its antimicrobial activity against multidrug-resistant Gram-negative bacteria[J]. CPT Pharmacometrics Syst Pharmacol, 2019, 8(11): 815-824.
NMPA. 藥物非临床药代动力学研究技术指导原则 [EB/OL]. https://www.cde.org.cn/zdyz/domesticinfopage?zdyzIdCODE=1f823fceeb386389432d22162290e61e.
Bhagwat S S, Periasamy H, Takalkar S S, et al. The novel β-lactam enhancer zidebactam augments the in vivo pharmacodynamic activity of cefepime in a neutropenic mouse lung Acinetobacter baumannii infection model[J]. Antimicrob Agents Chemother, 2019, 63(4): e02146-18.
Singh R, Kim A, Tanudra M A, et al. Pharmacokinetics/pharmacodynamics of a β-lactam and β-lactamase inhibitor combination: a novel approach for aztreonam/avibactam[J]. J Antimicrob Chemother, 2015, 70(9): 2618-2626.
Griffith D C, Sabet M, Tarazi Z, et al. Pharmacokinetics/pharmacodynamics of vaborbactam, a novel beta-lactamase inhibitor, in combination with meropenem[J]. Antimicrob Agents Chemother, 2018, 63(1): e01659-18.
Bhagunde P, Zhang Z, Racine F, et al. A translational pharmacokinetic/pharmacodynamic model to characterize bacterial kill in the presence of imipenem-relebactam[J]. Int J Infect Dis, 2019, 89: 55-61.
Coleman K, Levasseur P, Girard A M, et al. Activities of ceftazidime and avibactam against β-lactamase-producing Enterobacteriaceae in a hollow-fiber pharmacodynamic model[J]. Antimicrob Agents Chemother, 2014, 58(6): 3366-3372.
Berkhout J, Melchers Maria J, Van Mil Anita C, et al. Pharmacodynamics of ceftazidime and avibactam in neutropenic mice with thigh or lung infection[J]. Antimicrob Agents Chemother, 2016, 60(1): 368-375.
Wu J, Racine F, Wismer Michael K, et al. Exploring the pharmacokinetic/pharmacodynamic relationship of relebactam (MK-7655) in combination with imipenem in a hollow-fiber infection model[J]. Antimicrob Agents Chemother, 2018, 62(5): e02323-17.
基金项目:国家自然科学基金(No. 82173896)和上海市领军人才(No. LJ2016-01)
作者简介:卞星晨,女,生于1994年,主要研究方向为抗菌药物系统药理学研究,E-mail: bxc19940216@163.com
