富集纤维素降解菌群过程中微生物群落多样性分析
2023-03-27王彦伟祝其丽谭芙蓉胡国全何明雄高立洪
王彦伟,祝其丽,吴 波,谭芙蓉,胡国全,何明雄,高立洪
(1.农业部沼气科学研究所 秸秆资源化利用科技创新团队,四川 成都 610041;2.农业农村部农村可再生能源开发与利用重点实验室,四川 成都 610041;3.农业废弃物资源化利用技术与设备研发重点实验室,重庆 404100)
木质纤维素是地球上储量最大的可再生的生物质资源,但木质纤维素结构非常复杂,使其难以快速被微生物降解,这便限制了纤维素资源化的有效利用[1-3]。获得能高效转化纤维素的微生物是纤维素资源高值化利用的基础。利用微生物共培养(复合菌系)进行纤维素降解与生物转化是实现其高值化利用的有效途径。实现纤维素废弃物的高效利用,已成为缓解能源紧张、提高环境质量、促进经济和社会可持续发展的有效方法[4-5]。但微生物共培养体系复杂,不同种类的微生物如何在共培养体系发挥作用,仍然是急需解析的问题之一。
微生物在自然界中的纤维素降解中起主要作用,纤维素降解微生物很容易在不同的生境中找到,如土壤、农业废弃物、昆虫内脏、粪便和反刍动物的瘤胃等[6]。瘤胃是一种具有多种微生物种群的天然纤维素降解系统,通过微生物相互作用降解纤维素材料[7]。Xing[8]等报道牛瘤胃微生物促进小麦秸秆生物降解了96%~97% 的纤维素和半纤维素。Li[9]等在瘤中发现2个微生物组可以促进青储秸秆糖的转化。但是利用单一菌株进行木质纤维素降解时,由于底物抑制和反馈抑制作用的存在,使得木质纤维素降解水平低下[10-12]。近些年来,人们发现混合的菌群对复杂的有机污染物及天然难降解物质如多氯联苯、多环芳烃和合成燃料等有较强的降解能力[13-15]。大多数情况下,多菌的降解能力都远远高于单菌,因而纤维素降解复合菌系已越来越受到人们的重视。
复合菌系降解木质纤维素时能够避免单一菌株降解时产生的底物抑制和反馈抑制作用,因产酶更加多样,从而能够有效地降解木质纤维素。Kato[16]等通过研究复合菌系中主要菌株间的关系,从中分离主要菌株,并将厌氧的纤维素降解菌和好氧的非纤维素降解菌株组合在一起,发现非纤维素降解菌对纤维素降解起了非常重要的作用。本研究以连续传代的纤维素降解复合菌系为研究对象,利用16S rRNA 及ITS基因扩增子高通量测序方法,分析不同传代复合菌系降解纤维素过程中各微生物分类单元的群落组成及其相对丰度变化,探究复合菌系群落结构的动态变化规律。
1 材料与方法
1.1 材料
接种物:取自四川省金堂县农村合作社新鲜屠宰的山羊瘤胃内溶物,4℃保存于无菌的血清瓶中。配制赫奇逊无机盐培养基(g·L-1):KH2PO41.0,NaCl 0.1,MgSO4·7H2O 0.3,NaNO32.5,FeCl30.001,CaCl20.1,pH值7.2。DNeasy Power Soil Kit。
1.2 样品的传代培养
以羧甲基纤维素钠为唯一碳源,配制赫奇逊无机盐培养基,将培养基分装至三角瓶中,装液量为40%(体积比),1×105Pa、121℃灭菌15 min。接种底物为瘤胃内溶物,接种量为10%(体积比),30℃ 150 rmp·min-1摇床培养,连续传代培养,每隔6 d传一代,共传代20次,每一代取样保存至-80℃冰箱。
1.3 微生物组总DNA提取
选取20次样品中的10个样品,包含第一代及最后一代编号C1-C10,采用TIANamp公司的DNeasy PowerSoilKit进行抽提,采用荧光分光光度计(Quantifluor-ST fluorometer, Promega, E6090),在260 nm和280 nm处分别测定DNA的吸光值,检测DNA的浓度,并用1%的琼脂糖凝胶电泳检测DNA的质量。调整DNA溶液浓度,DNA工作液保存于4℃,储存液保存于-20℃。
1.4 PCR扩增
以上述DNA 为模板,利用引物对各样品基因组DNA V3~V4 区域及真菌ITS序列进行PCR扩增,将反应体系置于ABI Gene Amp®9700 型PCR 仪进行扩增,PCR产物参照电泳初步定量结果,将PCR 产物用QuantiFluorTM-ST 蓝色荧光定量系统(购于Promega公司)进行检测定量。利用TruSeqTM DNA Sample Prep Kit 回收纯化PCR 产物,Tris-HCl洗脱,2%琼脂糖凝胶电泳检测PCR产物,将扩增产物送至上海派森诺生物科技有限公司,采用Illumina MiSeq 测序平台进行测序,构建16S rRNA文库及ITS文库。
1.5 数据分析
首先对高通量测序的原始下机数据进行初步质量筛查;通过质量初筛的序列按照引物和Barcode信息,识别分配入对应样品,并去除嵌合体等疑问序列;对获得的序列进行OTU归并划分,每个OTU的代表序列用于分类地位鉴定以及系统发育学分析;根据OTU在不同样品中的丰度分布,评估每个样品的多样性水平,并通过稀疏曲线反映测序深度是否达标;对各样品(组)在不同分类水平的具体组成进行分析(并检验组间是否具有统计学差异);通过多种多变量统计学分析工具,进一步衡量不同样品(组)间的菌群结构差异及与差异相关的物种;根据物种在各样品中的组成分布,构建互作关联网络;根据16S rRNA及ITS基因测序结果,预测各样品的菌群代谢功能。
2 结果与分析
2.1 样品测序量
对10个连续传代样品中细菌16S rRNA序列测定,获得序列量在53885~67722之间,经过去除低质量、减噪等方法过滤,每个样品的有效序列在46442~59133条,平均测序覆盖率为99.2%。碱基长度分布于400~450之间。10个样品的真菌ITS序列测定序列量在 78340~97643之间,经过去除低质量、减噪等方法过滤后,每个样品的有效序列在71456~89874条,平均测序覆盖率为99.6%。碱基长度分布于100~300之间。
2.2 样品OUT分布情况
10个样品中细菌属水平测序比对结果丰富,其中样品OUT比对结果在262~535个属,随着传代次数增多,样品中细菌属逐渐减少,其中样品C1(第1代)测序属最多达到535,样品C8(第16代)测序属最少209可比对属,随着传代进行C8,C9,C10复合系逐渐稳定,细菌构建完成(见图1)。10个样品中真菌种水平序列比对结果丰富,其中OUT比对结果在96~139个种之间,随着传代次数增多,样品中真菌种逐渐增多,C10样品测序属种达到139个。C2样品中可比对种最少为96个。样品中真菌的可比对序列与细菌变化趋势不同(见图2)。
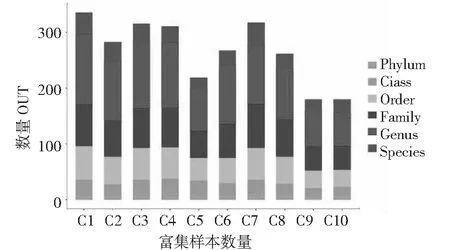
图1 细菌OTU分布
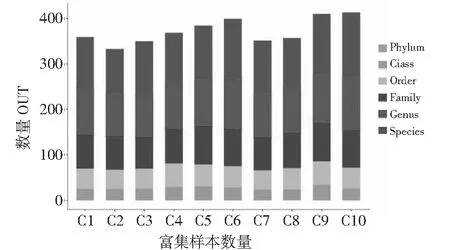
图2 真菌OTU分布
2.3 细菌,真菌群落丰富度和多样性的影响
为了能较为全面的评估10个传代样品中微生物群落的Alpha多样性,表1反映了10个样品的多样性差异,在细菌群落多样性分析中,传代后期样品C8,C9,C10的 Chao1及Observed species数值较小,表明丰富度较前7代样品差异显著,后期传代样品C8的Shannon和Simpson数值较小,多样性较其他样品差异显著,C8样品的Pielou’s e指数小,样品的均匀度较其他样品差异显著。前期样品的细菌丰富度、多样性及均匀度均好于后期传代样品。真菌多样性分析中,前期传代样品C1,C2及C3的Chao1及Observed species数值较小,表明丰富度较后7代样品差异显著。样品C10的Shannon和Simpson数值较小,多样性较其他样品差异显著。C10样品的Pielou’s e指数小,样品的均匀度较其他样品差异显著,真菌群落多样性分析中,前期传代样品中的丰富度较差,但是样品的多样性及均匀性高于后期传代样品。富集样品的前期丰度较高,后期变少,可能是部分菌群在富集筛选过程中退化,作用降低,优势菌群增加,丰度降低。

表1 样品Alpha多样性指数表
2.4 传代样品的微生物群落组成分析
对各个物种的组内样品丰度值进行归一化后,再求样品平均值,分析10个细菌属水平,所展示样品前20的细菌属如图3,10个样品中各种属展示如下。德沃斯氏菌属(Devosiasp.)占13%,纤维单胞菌属(Cellulomonassp.)占12%,异根瘤菌属(Allorhizobium)-新根瘤菌属(Neorhizobium)-副根瘤菌属(Pararhizobium)-根瘤菌属(Rhizobium)占4%。在前期样品(C1)中链霉菌属(Streptomycessp.)大量存在,这可能与瘤胃液的微生物类群相关,随着传代的进行,德沃斯氏菌属(Devosiasp.)、纤维单胞菌属(Cellulomonassp.)逐渐成为优势菌群。其中在后期传代样品(C7)中纤维弧菌属(Cellvibriosp.)、砂单胞菌属(Arenimonassp.)占据了优势地位,有别于其他后期样品。C8、C9和C10样品中群落结构趋于稳定。结果表明:厚壁菌门中的种属占优势地位,这和报道中的瘤胃微生物相似,牛瘤胃中厚壁菌门也是秸秆降解的优势种群[17-18],同时在测序分析中仍然有大量的微生物未被鉴定到属。已有的研究也表明:自然资源中仍然有许多未被发现的微生物,可能具有重要的生物学功能。
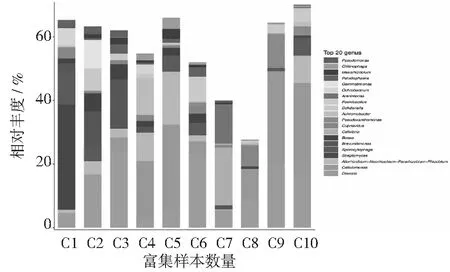
图3 样品细菌属分类水平物种组成
对各个物种的组内样品丰度值进行归一化后,再求样品平均值,分析10个样品真菌属水平,所展示样品前20的真菌属如图4所示,其中曲霉属(Aspergillussp.)占8%,古根霉属(Archaeorhizomycessp.)占6%,被孢霉属(Mortierellasp.)占3%等。未鉴定到属分类的真菌菌属占比最高,大量的未培养的真菌仍然需要进一步挖掘。
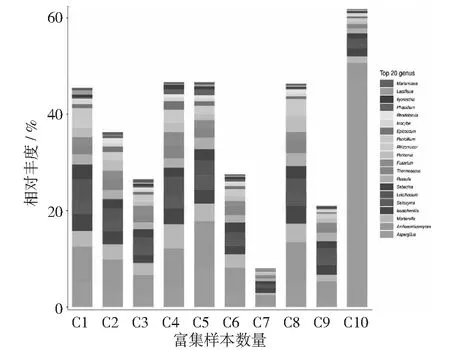
图4 样品真菌属分类水平物种组成
2.5 组间差异
为了比较样品间的物种组成差异,实现对各样品的物种丰度分布趋势的展示,研究中,使用属水平的分类单元组成作为分析对象,使用平均丰度前50位的属的丰度数据绘制热图。细菌属热图分析不同样品的差异较为显著,不同传代样品之间优势种群存在显著差异,纤维弧菌属(Cellvibriosp.)和 砂单胞菌属(Arenimonassp.)仅在C7样品中较为丰富,纤维单胞菌属(Cellulomonassp.)和假黄色单胞菌属(Pseudoxanthomonassp.)在C4、C9、C10样品中较为丰富,在C1、C2、C3样品中Pseudomonassp属占有优势,但随着传代的进行,在后面的传代样品中,逐渐被其他优势菌群替代。类芽孢杆菌属(Paenibacillussp.)和 戈马单胞菌属(Gommatimonassp.)仅在C2中占有优势。德沃斯氏菌属(Devosiasp.)和独岛杆菌属(Dokdonellasp.)分别在C6、C9、C10样品中显示为优势菌属。热图分析显示随着传代的进行,样品中的细菌微生物群落逐渐改变,优势菌群发生变化,样品中的群落结构不稳定。
为了比较样品间的物种组成差异,实现对各样品的物种丰度分布趋势的展示,研究中,使用属水平的分类单元组成作为分析对象,使用平均丰度前50位的属的丰度数据绘制热图。真菌属热图分析表明不同样品的差异较为显著,C10样品中仅存在曲霉属(Aspergillussp.)优势种群。C1、C2、C3、C4、C5样品中优势菌种较多,分别是Lactiflcussp.、peniollumsp.、红酵母菌属(Rhodotorulasp.)、Llyonectriasp.、附球菌属(Epicoccumsp.)、星裂盘菌属(phacidiumsp.)。C7,C9样品中优势菌群未被发掘,在C8样品中黑团胞属(Periconiasp.)占有优势。
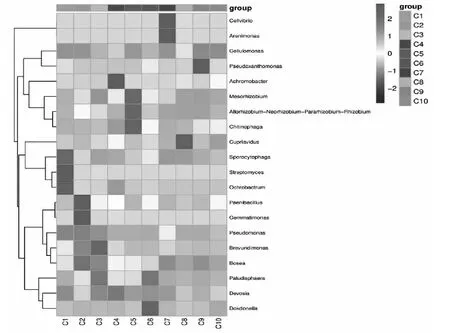
图5 细菌属水平hotmap 分析
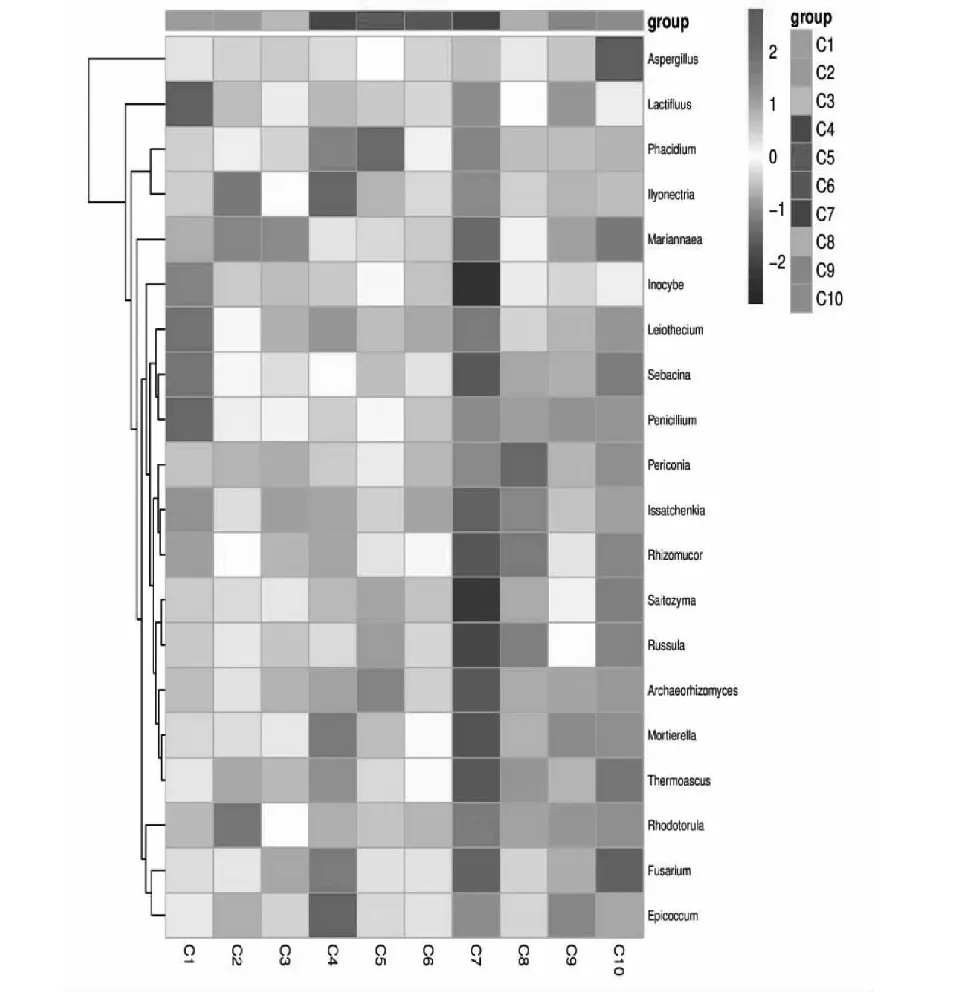
图6 真菌属水平heatmap分析
3 讨论
本研究采用Illumina MiSeq 技术对连续传代的复合系样品的群落进行高通量测序,组间差异性分析显示不同传代样品微生物群落结构存在明显的差异,细菌,真菌热图分析表明不同传代样品中的细菌,真菌的优势菌群不同,前期传代样品(C1,C2,C3,C4和C5)中的假单胞菌属(Pseudomonassp.)、生孢噬纤维菌属(Sporocytophagasp.)、链霉菌属(Streptomycessp.)、苍白杆菌属(Ochrobactrumsp.)、类芽孢杆菌属(Paenibacillussp.),gemmatatimonassp.、短波单胞菌属(Brevundimonassp.)、无色杆菌属(Achromobactersp.)、中慢生根瘤菌属(Mesorhizobiumsp.)、鞘氨醇菌属(Chitinophagasp.)等优势种属逐渐消失,前期传代样品中的优势细菌属逐渐被后期传代样品中的独岛杆菌属(Dokdonellasp.)、纤维弧菌属(Cellvibriosp.)、假黄色单胞菌属(Pseudoxanthomonassp.)等取代,前期传代样品(C1,C2,C3,C4,C5)中优势真菌Lactifluussp.、青霉属(Peniollumsp.)、llyonectriasp.等被黑团胞属(Periconiasp.)、曲霉属(Aspergillussp.)取代。以CMCNa为唯一碳源连续富集、限制性培养过程中,菌群结构一直处于变化的状态,使随机性过程增加。后期传代样品优势细菌纤维弧菌属(Cellvibriosp.)和假黄色单胞菌属(Pseudoxanthomonassp.)都被报道过具有纤维素降解能力的菌属[19-22],这类菌属的保留,可能与其具有木质纤维素分解功能的或者辅助降解纤维素功能有关。测序结果分析表明复合菌系中存在与纤维素降解有直接关系的菌,也存在可能有未知的功能,或者因适应培养基选择并保留,也可能在接下来的传代培养中被“淘汰”。
通过分析10个样品中的OUT分类,在细菌分类中unclassified_Microbacteriaceae,德沃斯氏菌属(Devosiasp.)和纤维单胞菌属(Cellulomonassp.)3种类型的种属占比较高,其中德沃斯氏菌属(Devosiasp.)被报道过存在于污泥秸秆堆肥体系、棉花秸秆堆肥体系和秸秆还田的土壤等不同的秸秆降解体系中[23-24],其在秸秆降解复合菌系中发挥着重要的作用。纤维单胞菌属(Cellulomonassp.)[25-26]是细菌属中纤维素酶活力较强的菌株,该菌属可通过分泌几种胞外纤维素酶对秸秆进行水解,在细菌纤维素降解过程中有重要的研究意义。在真菌OUT分类中unclassified_fungi(49%),未鉴定到属的真菌占大部分,其作用有待于进一步解析。Aspergillussp.(8%)[27-28]及古根霉属(Archaeorhizomycessp.)占6%[29-30]都具备降解纤维素的能力,在秸秆堆肥富集物中都被监测到是优势菌群,被孢霉属(Mortierellasp)占3%在复合系中的作用不明。在自然环境中,木质纤维素的降解是多种微生物共同作用的结晶,纤维素的彻底降解是通过多种微生物长时间相互作用的结果。对于纯培养的单菌,不论是细菌还是真菌,很少有明确报道表明单菌具有较强的天然木质纤维素分解能力。
以CMCNa为底物富集纤维素降解菌系,测序分析表明复合系中存在大量的纤维素降解菌,在选择性底物的作用下,特定的微生物得到了富集,但是连续传代的样品并没有获得稳定的复合系,这可能和培养时间、培养基组成等有关。要想获得稳定的复合系,还需要进一步优化培养条件等。
