CRISPR-Cas9基因编辑技术对细胞内源蛋白进行荧光标记的实验操作
2023-03-03吴仲胜高誉杜勇涛党颂何康敏
吴仲胜,高誉,杜勇涛,党颂,何康敏
实验操作指南
CRISPR-Cas9基因编辑技术对细胞内源蛋白进行荧光标记的实验操作
吴仲胜1,2,高誉1,2,杜勇涛1,2,党颂1,何康敏1,2
1. 中国科学院遗传与发育生物学研究所,分子发育生物学国家重点实验室,北京 100101 2. 中国科学院大学,北京 100049
CRISPR-Cas9是目前广泛应用的基因编辑技术,可对目的基因进行高效精准编辑,快速实现目的基因的敲除或敲入。Cas9蛋白在sgRNA引导下对靶序列进行剪切并造成DNA双链断裂,在与剪切位点两端同源的DNA模板序列存在时,可通过同源重组修复方式引入外源序列,实现荧光蛋白或其他标签在基因组上的精准敲入,进而实现对内源蛋白进行荧光标签的融合标记。通过基因编辑技术对内源目的蛋白进行标记,可避免由于过表达造成蛋白质定位、动力学或功能等的潜在影响,可显著提升细胞成像实验的稳定性和可重复性。本文重点介绍了利用CRISPR-Cas9基因编辑系统对目的蛋白进行荧光蛋白或自标记蛋白标签标记的方法与操作流程,为构建内源蛋白荧光标记的哺乳动物细胞系提供参考。
CRISPR-Cas9基因编辑;活细胞;内源蛋白;荧光蛋白;自标记蛋白标签
蛋白质的定位与功能密切相关。通过对各种蛋白质在细胞内特别是活细胞内的定位、动力学、分子间互作等进行精准表征和分析,进而研究蛋白质的功能和调控机制等具有重要意义。目前已经发展了多种在活细胞内对目的蛋白进行荧光标记的方法,包括荧光蛋白(fluorescent protein)、自标记蛋白标签(self-labeling protein tag)、荧光共价标记的配体或多肽、非天然氨基酸标记等[1,2]。荧光蛋白标记是最常用的可遗传编码的蛋白质标记方法,目前已经发展了多种荧光强度和光稳定性质优异的荧光蛋白,其荧光发射光谱实现了从蓝到红外光谱的覆盖,可基本满足激光共聚焦显微镜成像、单分子成像和超分辨率成像等不同需求和成像目的[3,4]。HaloTag等自标记蛋白标签的标记,是另外一种广泛应用的可遗传编码的活细胞蛋白标记方法,通过与相应荧光染料标记的配体进行共价反应,实现对目的蛋白进行特异荧光标记[5,6]。对目的蛋白进行荧光蛋白或自标记蛋白标签的融合表达,可通过质粒瞬时外源过表达、随机插入基因组的稳定过表达或者基因编辑精准插入目的基因等方式实现。CRISPR-Cas9介导的基因编辑体系,通过对目的基因进行精准剪切,在含有荧光蛋白或自标记标签蛋白cDNA序列的修复模板的介导下,采用同源重组的方式在目的基因上精准敲入(knock-in)荧光标签的DNA序列,进而实现对内源目的蛋白的荧光标记[7]。通过基因编辑技术对内源蛋白进行荧光标记,可有效避免蛋白质过表达可能造成目的蛋白的定位或功能的异常。此外,基因编辑细胞具有更好的均一性和稳定性,有效提升了细胞成像实验的可重复性,也更有利于对图像进行定量分析。目前已经发展了多种不同的基因编辑方法和策略,实现对目的蛋白的荧光蛋白或自标记蛋白标签的标记[8~10],本文重点阐述了利用CRISPR-Cas9基因编辑系统,对哺乳动物细胞特定目的蛋白进行荧光蛋白或自标记蛋白标签标记的方法与操作流程。
1 CRISPR-Cas9基因编辑knock-in实验设计
1.1 荧光标签的选择
目前已经发展了多种荧光蛋白及变体,包括自身发光的荧光蛋白如EGFP、拆分荧光蛋白(split fluorescent protein)[11,12]、二聚化依赖荧光蛋白[13]和光激活/光转化荧光蛋白[14]等。常用的自发光绿色荧光蛋白有EGFP[15]、mEGFP[16]和mNeonGreen[17],常用的红色自发光荧光蛋白有mCherry[18]、TagRFP[19]以及新近发展的mScarlet/mScarlet-H/ mScarlet-I[20]以及FusionRed-MQV等[21]。其中mNeonGreen、mScarlet-I和FusionRed-MQV等具有较高的量子产率和荧光强度且均为单体,适合于活细胞成像和追踪。FPbase网站(https://www.fpbase. org/)收录了目前发表的各种荧光蛋白的详细信息,可对各种荧光蛋白的序列、光学和物理化学性质等进行查询和比较。
拆分荧光蛋白如拆分GFP (split GFP)是将GFP拆分为GFP11(β-折叠链11,残基215~230)和GFP1-10(β-折叠链1~10,残基1~214)两个片段,其中GFP11用柔性linker连接到目的蛋白上,GFP1-10单独表达。GFP11和GFP1-10单独存在时均不发光,当共存时会自组装成为完整可发光的GFP分子[11]。该系统的优势是GFP11片段较小,降低了对目的蛋白的影响,此外也减少了用于同源重组的模板的长度,使得高通量荧光标记细胞内源蛋白成为可能[22]。该系统的另一个优势是降低了背景荧光。
自标记蛋白标签如HaloTag[23]、SNAP-tag[24,25]和CLIP-tag[26],是近些年使用逐渐广泛的一类荧光标记工具。其自身不发光,但通过与荧光染料标记的配体进行共价反应,可对目的蛋白进行荧光染料的标记。新近发展的Janelia Fluor系列荧光染料,具有亮度高、光稳定性高和可穿过细胞膜等优异性质,适合用于活细胞膜及细胞内标签蛋白的荧光成像和动态追踪,也十分适合于活细胞单分子成像和追踪[27]。此外,通过选择结合不同激发/发射波长荧光染料的配体,可对目的蛋白进行不同颜色荧光的标记,进而实现多色活细胞成像的目的。
1.2 荧光标签插入位置以及linker的选择
荧光标签插入目的蛋白的位置,可位于其N端、C端或中间,具体由该蛋白质自身的结构和功能等性质决定。通常情况下,通过调研已发表文献或数据库中蛋白结构等,或者在瞬时转染质粒上进行多种插入位点尝试,以确保荧光标签的插入不会对目的蛋白的定位、动力学和功能造成影响。此外,为了保证目的蛋白和荧光标签蛋白的正确折叠以及减少荧光标签对目的蛋白结构和功能的影响,通常会在目的蛋白和荧光标签之间插入一段linker蛋白序列,如常用的富含甘氨酸的柔性linker如(GGGGS)3[28]和(GGS)n[29]。在本实验操作流程中选取(GGS)3作为融合蛋白的linker。
1.3 CRISPR-Cas9体系的选择
在CRISPR-Cas9系统中,Cas9 (SpCas9)蛋白在gRNA (guide RNA)的引导下完成对DNA的定点切割。Cas9蛋白行使功能需crRNA (crispr RNA)与tracrRNA (trans-activating crRNA)共同参与[30]。通过将crRNA和tracrRNA融合形成sgRNA (single guide RNA),而后在sgRNA的引导下,Cas9酶定位于特定DNA序列上,进行DNA双链切割和对目的基因进行编辑[7](图1)。在利用CRISPR-Cas9体系对靶细胞进行knock-in基因编辑时,其体系成分具有多种选择,但最终目的是将Cas9核酸酶(可为质粒、mRNA、重组蛋白、或者已经稳定表达Cas9的细胞系等)、sgRNA (可为质粒或包含sgRNA的DNA片段等)、同源重组修复模板(可为质粒、单链DNA或者双链DNA等)递送进入细胞内[31]。
在本实验流程中,将以溶酶体的标志分子LAMP1蛋白为例进行实验设计和流程介绍。本文分别选择了红色荧光蛋白mScarlet-I和HaloTag为荧光标签,选择的linker为(GGS)3。编码mScarlet-I和HaloTag的cDNA长度分别为696 bp和891 bp,将以pUC19 (Addgene plasmid #50005)为载体构建同源重组修复模板(以下称之为供体质粒)[32,33]。考虑到实验的简捷和稳定性,本实验流程利用pSpCas9(BB)-2A-Puro (Addgene plasmid #62988)或pSpCas9(BB)-2A-GFP (Addgene plasmid #48138)质粒递送Cas9和sgRNA。
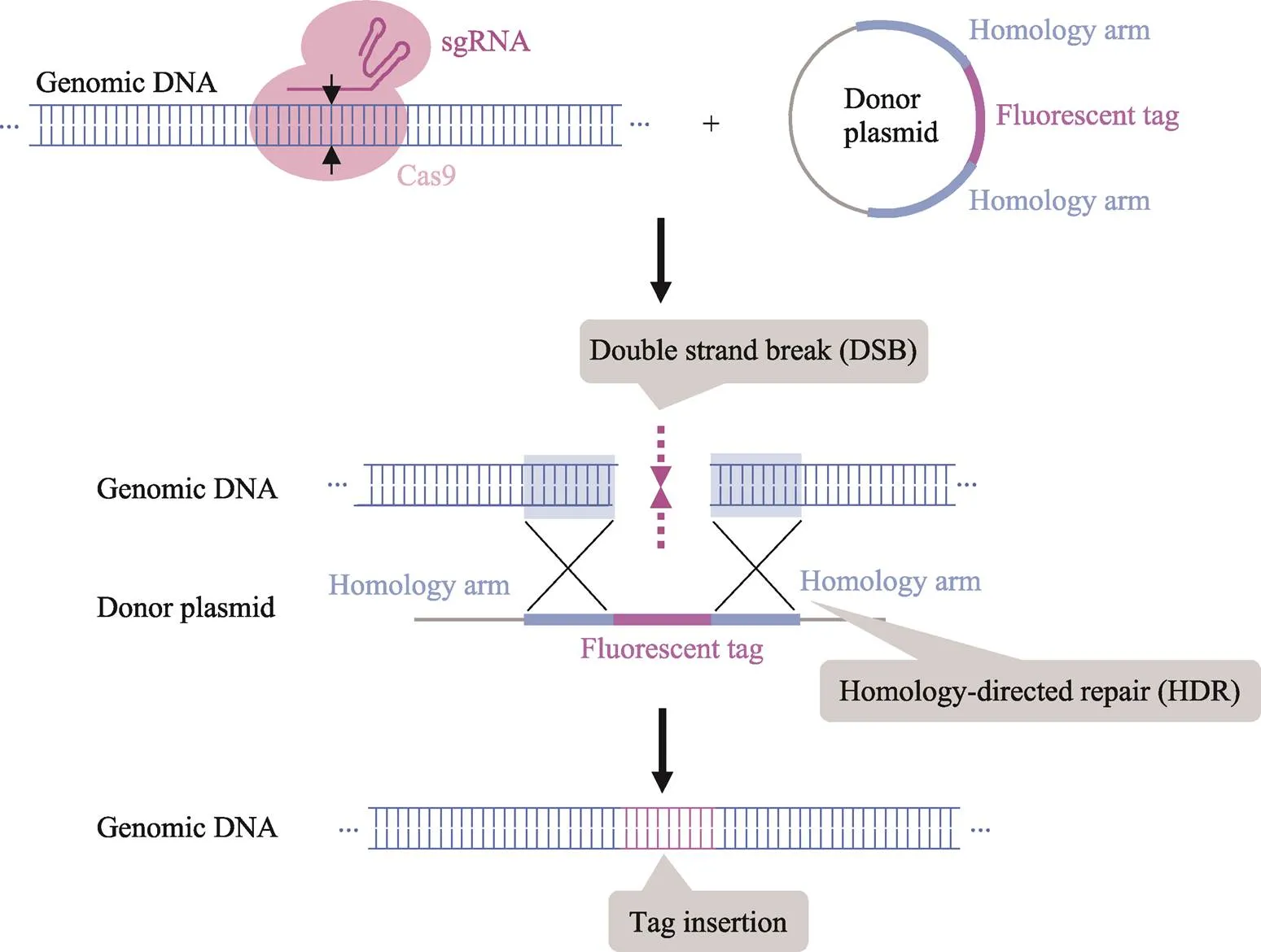
图1 利用CRISPR-Cas9基因编辑技术进行荧光标签基因敲入的示意图
1.4 同源重组供体质粒的设计
供体质粒对同源重组的效率具有重要的影响,在实验过程中需要注意以下几点:
1.4.1 目的基因的基因组DNA (genomic DNA, gDNA)和CDS (Coding DNA Sequence)序列的查找:根据目的基因种属、基因名称、转录本(transcript variant)等信息,在Ensembl、NCBI或者其他网站进行查询。
1.4.2 插入位点的确定:在确定好荧光标签插入目的蛋白的位置后,还应注意DNA双链断裂处与插入位点之间的距离不应超过100 bp,理想的距离是在10 bp以内,这样可以最大限度地提高同源重组修复的效率[34]。
1.4.3 供体质粒的设计:供体质粒内包含了三段拼接的DNA序列,分别是(1)目的基因插入位点上游的DNA序列、(2)插入标签的DNA序列,(3)目的基因插入位点下游的DNA序列。若插入的是荧光蛋白或自标记蛋白标签,通常选取插入位点上下游长度约为600~800 bp作为两侧的同源臂序列。以靶细胞的基因组为模板,利用PCR扩增上下游同源臂的序列,亦可直接合成。
在本实验流程中,通过查阅文献以及基于瞬时转染质粒的测试结果,将荧光标签插入到目的基因的C端。由于荧光蛋白mScarlet-I和自标记蛋白标签HaloTag的基因片段较长,供体质粒包含的同源臂为的gDNA终止密码子上下游各约800 bp。同源修复的供体质粒设计如图2A和2B所示。
1.5 sgRNA序列设计
sgRNA在CRISPR-Cas9系统发挥着引导作用,对knock-in的特异性和效率至关重要。可借助“CRISPOR”[35]和“CRISPick”[36]等网站设计sgRNA,选取特异性和效率俱佳的序列。sgRNA的靶向序列可以位于正义链,也可以位于反义链,但sgRNA的靶向序列应避免4个以上的T结尾,以防止形成终止转录信号,且GC含量最佳为40%~60%。为了尽可能提高实验效率和减少脱靶效应,可挑选切割效率高、特异性好的两条或以上的sgRNA同时测试和开展后续实验。
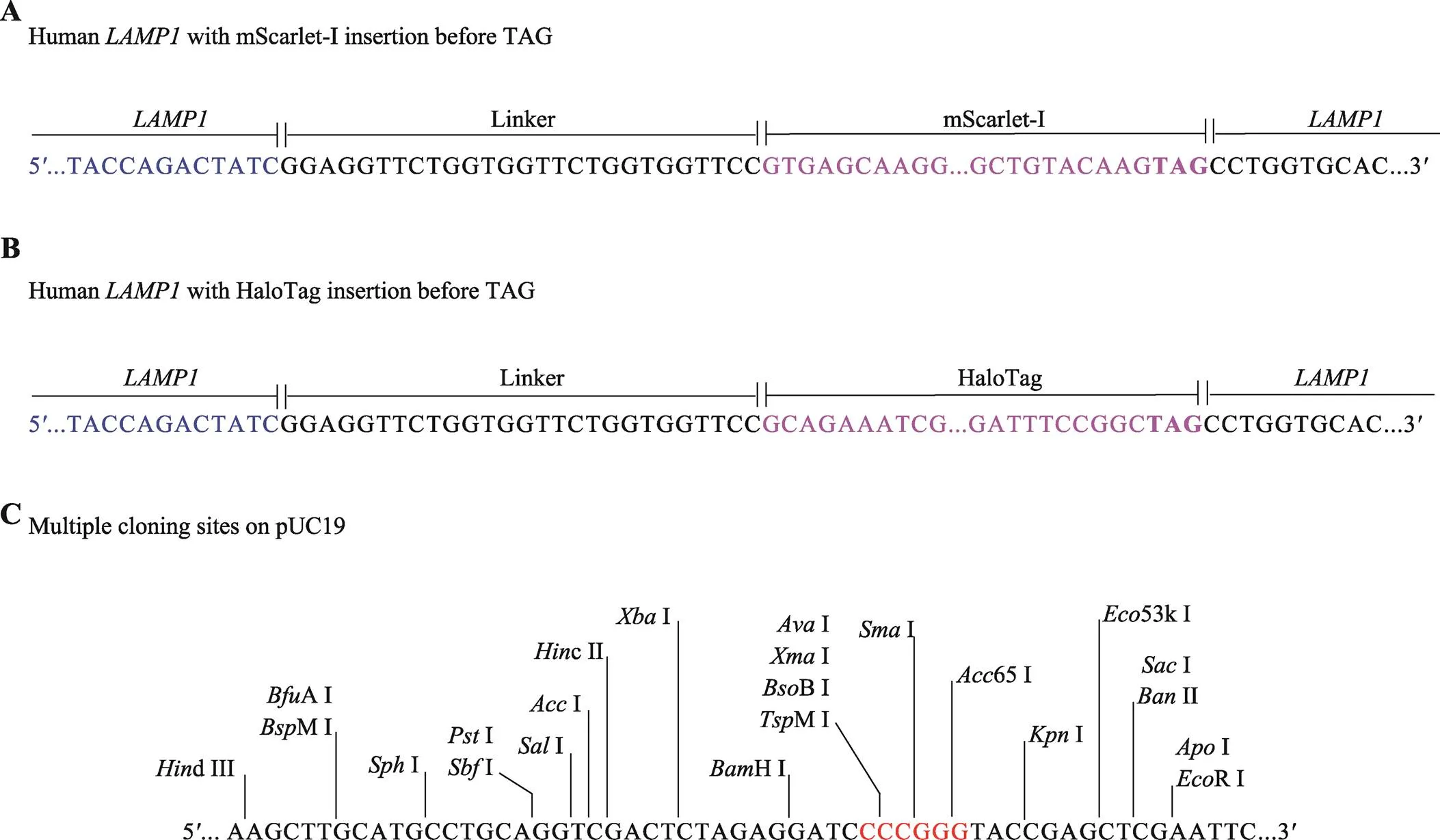
图2 利用CRISPR-Cas9基因编辑技术将mScarlet-I或HaloTag序列插入至LAMP1基因C端的设计
A: 基因编辑表达LAMP1-mScarlet-I的序列设计;B: 基因编辑表达LAMP1-Halo的序列设计;C: pUC19载体的多克隆位点。
2 试剂与材料
实验相关的主要试剂与耗材信息详见表1。
3 基因编辑相关质粒的构建
3.1 供体质粒的构建
3.1.1 gDNA的提取:将培养皿中的细胞消化并收集后,使用TIANamp Genomic DNA试剂盒提取gDNA,保存于4℃或–20℃以备后续使用。
3.1.2 线性化质粒的制备:选择合适的限制性内切酶如I在模板质粒pUC19的多克隆位点(图2C)处进行酶切,胶回收后得到线性化的质粒。
3.1.3 三段DNA序列的制备:荧光蛋白的编码序列以已含有该序列的质粒为模板,上下游同源臂的碱基序列可直接合成,也可以3.1.1步骤制备的gDNA为模板,经PCR扩增和胶回收处理后得到上下游同源臂的DNA片段。需要注意的是,这三段DNA序列彼此连接的区域以及与质粒连接的区域存在一个片段大小为18~30 bp的同源臂,该片段是通过设计相应的引物进行PCR扩增后添加的。PCR引物的Tm值以56~65℃为宜。
表1 试剂与耗材信息
3.1.4 线性化质粒和DNA片段的连接:利用pEASY-Uni Seamless Cloning and Assembly Kit将3.1.3制备的DNA片段和线性化的pUC19载体进行同源重组,得到包含荧光蛋白的供体质粒。建议连接时线性化载体与三个DNA片段加入量的摩尔比≥1∶3,三个DNA片段间的摩尔比为1∶1。分子质量摩尔数的转换可借助在线工具NEBiocalculator (https://nebiocalculator.neb.com/#!/ligation)进行测算。
3.1.5 转化和克隆筛选:按常规方法对连接产物进行转化,37℃培养12~14 h后,挑取单个菌落至液体培养基中扩大培养后,经测序鉴定后获得插入序列正确的供体质粒。
3.2 sgRNA质粒的构建
本实验流程采用的是美国麻省理工学院张峰实验室开发的pSpCas9(BB)-2A-Puro或pSpCas9(BB)- 2A-GFP载体,引物设计和详细操作步骤可参考Target Sequence Cloning Protocol (https://media.addgene.org/data/plasmids/62/62988/62988-attachment_KsK1asO9w4owD8K6wp8.pdf)。
3.2.1 线性载体的制备:用限制性核酸内切酶将pSpCas9(BB)-2A-Puro或pSpCas9(BB)-2A-GFP质粒进行酶切与去磷酸化,经琼脂糖凝胶电泳鉴定和胶回收后得到线性化的质粒。
3.2.2 sgRNA寡核苷酸双链的制备:根据选择的sgRNA,在引物合成时,正向寡核苷酸的5′端加上粘性末端CACCG,同时在反向寡核苷酸的3′端加上碱基C和5′端加上粘性末端CAAA。引物合成后,上下游引物在T4多聚核苷酸激酶的作用下后经梯度降温后得到sgRNA寡核苷酸双链。
3.2.3 sgRNA寡核苷酸双链和线性化质粒的连接:将得到的sgRNA寡核苷酸双链加入3.2.1步骤制备的线性化质粒中,在连接酶的作用下得到包含sgRNA的重组质粒。按常规方法对连接产物进行转化和培养,挑取单个菌落至液体培养基中扩大培养后,经测序鉴定后得到含有正确sgRNA序列的质粒。
3.3 质粒提取
建议使用可有效去除内毒素的质粒提取试剂盒提取供体质粒和sgRNA质粒,如利用NucleoBond Xtra Midi Plus试剂盒进行质粒提取。
4 细胞转染、分选和鉴定
在本实验流程中,使用CRISPR-Cas9基因编辑技术对内源蛋白进行荧光标记的实验主要包括以下三个步骤:(1)细胞转染、(2)阳性细胞分选和富集、(3)单克隆细胞的鉴定(图3)。
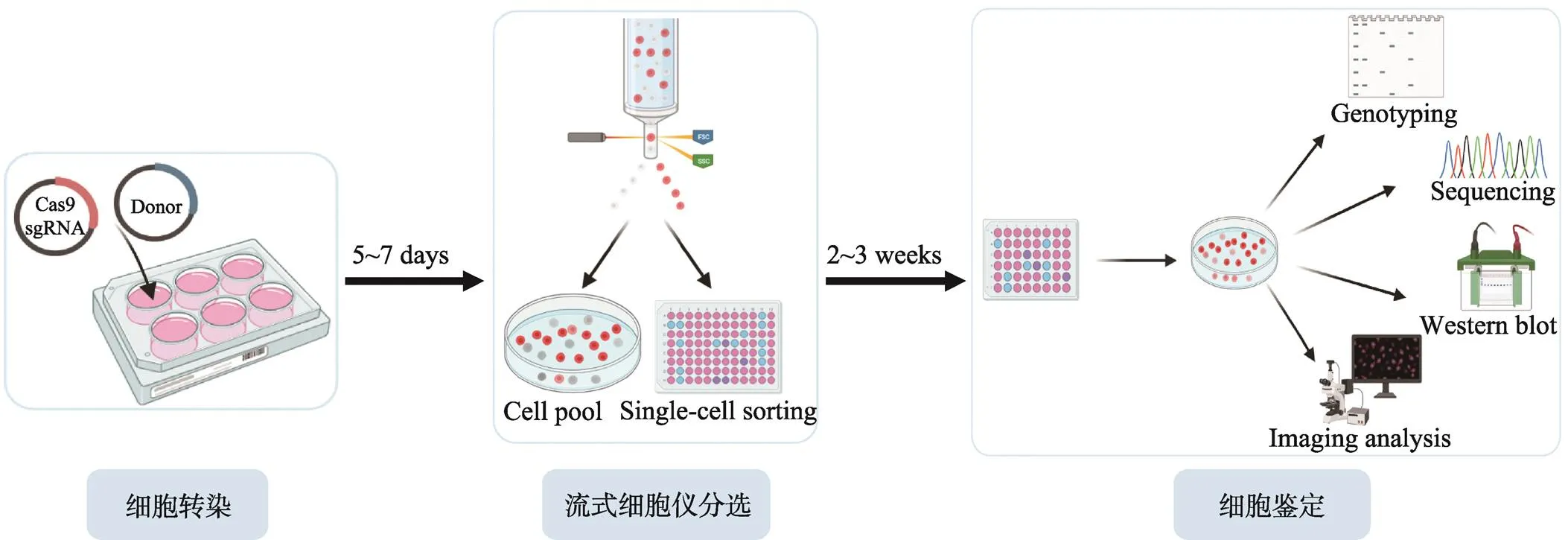
图3 基因编辑细胞系构建的实验流程
4.1 细胞转染
细胞复苏传代一次后,转染前16~24 h将细胞种在6孔培养板中,以转染时细胞密度为70%~80%为宜。按照Lipofectamine™ 3000试剂说明书,加入适量供体质粒和sgRNA质粒进行转染(每孔推荐质粒各600~800 ng)。8 h后更换新鲜培养基,24 h后传代至T75培养瓶中进行扩大培养。
4.2 细胞分选和富集
4.2.1 基因编辑阳性细胞的筛选方式:通常使用荧光(如荧光蛋白和荧光标记后的自标记蛋白标签)或利用嘌呤霉素(puromycin)等药物等对基因编辑的阳性细胞进行筛选和富集。若使用的供体质粒带有荧光蛋白或自标记蛋白标签,则在转染后5~7天,利用流式细胞仪分选并富集带有荧光的阳性细胞;若供体质粒不携带荧光标签,但sgRNA质粒带有荧光标签时,则在转染24 h后,利用流式细胞仪分选并富集带有荧光的阳性细胞。对于本实验流程中构建的基因编辑细胞系,其供体质粒包含有红色荧光蛋白mScarlet-I或者HaloTag,因此采用流式细胞仪对表达了mScarlet-I或者经荧光标记HaloTag配体染色后的细胞进行富集。
4.2.2 细胞分选样品准备:将转染后培养了5~7天的细胞进行消化并离心收集,而后加入适量无酚红的基础培养基如a-MEM进行重悬,经过45 μm孔径的细胞筛过滤后(去除细胞团)收集于流式管中。对于转染了LAMP1-HaloTag供体质粒的细胞,则需要在胰酶消化前进行染色。例如可使用Janelia Fluor 549 HaloTag配体对细胞进行10~20 min染色(终浓度10~20 nmol/L),使用PBS洗3遍后,再对其进行消化并离心收集等上述步骤。
4.2.3 阳性细胞分选和富集: 在利用流式细胞仪进行阳性细胞分选时,首先利用未转染的细胞设置荧光阴性的对照区域,以此为基础判断荧光信号阳性区域。根据目的蛋白的不同,首次经过流式细胞仪筛选的基因编辑细胞系,阳性率约为0.5%~5%左右,每次收集的阳性细胞数量可在20,000~50,000左右,随后将收集于流式管中的阳性细胞转移到48孔板中进行培养,当细胞密度达到95%~100%后传代至6孔板中进行扩大培养。
注意:在传代过程中可将少许细胞接种在成像皿中,进行首次成像鉴定,判断基因编辑细胞的阳性率以及目的蛋白的表达和分布是否正确。若阳性率低于10%,可对阳性细胞扩大培养后进行再次的分选富集;若阳性率较高且目的蛋白的表达和分布正确,可将单细胞分选至96孔板进行培养;若首次成像鉴定发现目的蛋白的表达和定位异常,则停止分选并调整实验设计。
4.2.4 单克隆细胞分选:根据实验需求,可选择构建单克隆细胞,也可通过多次分选获得高阳性率的细胞群。单克隆细胞系具有单一和稳定的优点,而对于某些特殊的不适合单克隆生长的细胞系,可通过分选收集阳性细胞群。在本实验流程介绍中,希望获得基因编辑单克隆细胞系,因此对荧光标记的LAMP1细胞系进行了单克隆分选。在分选时,流式细胞仪中mScarlet-I阳性细胞的信号区域分为高、中和低信号区域,分别代表了mScarlet-I纯和敲入、杂合敲入以及未敲入的细胞群。为获得mScarlet-I纯和敲入的细胞系,将高信号区的阳性细胞,利用流式细胞仪的单细胞分选方式分选至96孔板中继续培养(图4)。
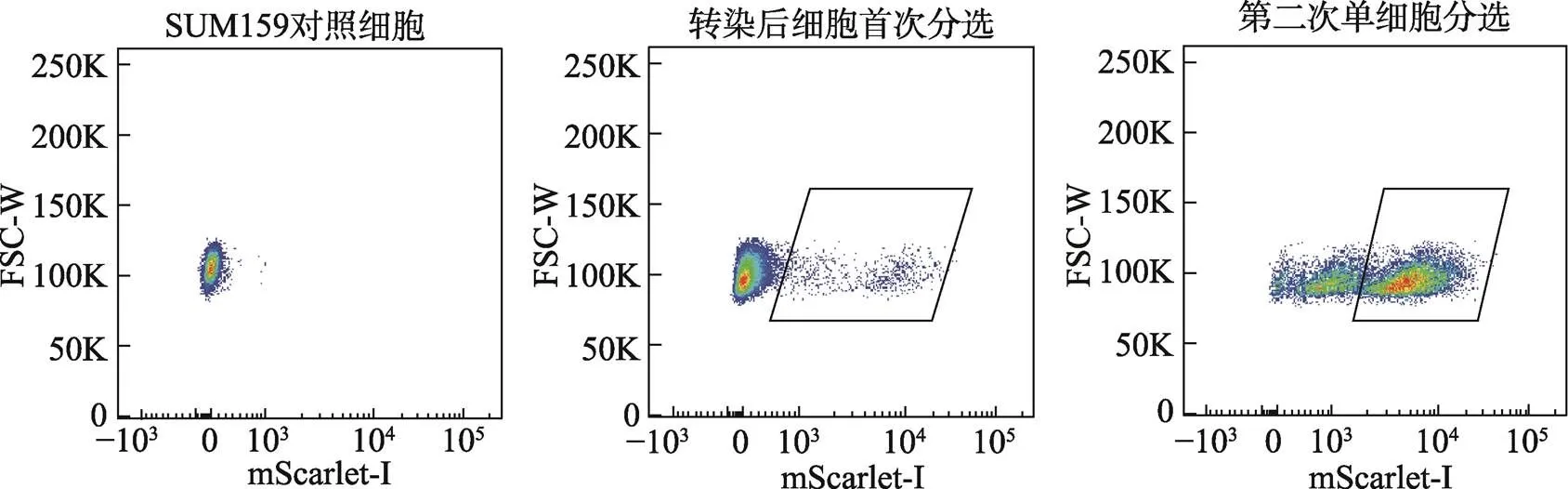
图4 利用流式细胞仪分选富集基因编辑表达LAMP1-mScarlet-I的细胞
5 单克隆细胞系的鉴定
5.1 单克隆细胞的初步筛选
分选在96孔板中的单克隆细胞,经过2~3周的培养后,在普通倒置显微镜的4倍物镜下可看到明显的细胞团。选择生长面积已经占据孔面积1/3及以上的单个细胞团,经胰酶消化和培养基中和后,对每个孔中的细胞进行吹打混匀并将其中1/2至2/3细胞传代至48孔板中继续培养,其余细胞转移至96孔PCR板中相应位置,离心(2000×, 室温10 min)后甩去液体,并倒扣于吸水纸上5 s以除去残留液体,随后每孔中加入10 μL QuickExtract DNA提取试剂并上下吹打30次重悬细胞,盖上胶膜后置于PCR仪中进行细胞裂解和gDNA获取,反应结束后将gDNA置于4 ℃或者–20 ℃长期保存。
5.2 PCR鉴定纯合和杂合单克隆细胞系
5.2.1 基因型鉴定引物设计:本流程示例中使用的人乳腺癌细胞系SUM159为二倍体细胞,因此对目的基因的荧光标签敲入会得到纯合和杂合两种类型。由于本示例中是将mScarlet-I插入到目的基因的C端,因此在目的基因的gDNA的终止密码子处上下游各选取400~500 bp左右的序列(不包含荧光蛋白),将这段约800~1000 bp的gDNA序列复制并粘贴至NCBI-Blast-Primer-blast (https://www.ncbi.nlm.nih.gov/tools/primer-blast/),按照设置(PCR片段大小:350~700 bp;Database:Refseg representative genomes)并生成引物,挑选其中特异性较好的2对引物进行后续测试。
5.2.2 基因型鉴定引物的PCR条件测试:由于需要单克隆鉴定的样品数量一般较多,同时对gDNA进行PCR有一定的失败率,因此可先对PCR引物和条件进行测试。对基因鉴定引物的退火温度进行梯度温度测试,同时设置阴性对照和阳性对照。PCR后进行凝胶电泳,根据条带的大小位置以及特异性等特征选择合适的引物和退火温度。
5.2.3 基因鉴定结果分析:利用预实验中得到的合适的PCR引物和条件,对挑选在96孔PCR板中的gDNA使用GoTaq酶进行PCR鉴定。以在SUM159细胞上基因编辑表达LAMP1-mScarlet-I为例,通常会得到三种PCR结果(图5A):(1)未实现基因编辑的单克隆的PCR产物,只有分子量较小的一条带(设计大小为403 bp);(2)单等位基因敲入(杂合)单克隆的PCR产物有两条带,其中一条分子量小(设计大小为403 bp)的条带为未插入mScarlet-I序列的PCR产物,另一条分子量较大的条带为插入了mScarlet-I序列的PCR产物(设计大小为1126 bp);(3)双等位基因敲入(纯和)只有一条分子量较大的条带(设计大小为1126 bp)。
5.3 PCR鉴定荧光标签插入位点的准确性
对于5.2得到的阳性克隆(纯合或者杂合单克隆细胞系),还需要判断荧光标签的cDNA是否插入了目的基因的正确位置。通常会设计检测插入位置的引物进行PCR鉴定,其中一条引物位于荧光蛋白的N或者C端,另外一条引物位于插入位置900~ 1200 bp之外的gDNA上(位于同源重组模板序列之外)。在本示例中,正向引物位于mScarlet-I的序列的C末端附近,而反向引物位于在终止密码子的下游约900~1200 bp。经过PCR扩增和凝胶电泳后,判断是否有条带以及得到的电泳条带片段与预期大小是否相符。若扩增出电泳条带且片段大小符合预期,则可初步判定插入的荧光标签序列的位置位于目的基因的相应位置(图5B),对这些克隆可进一步测序鉴定插入是否准确。
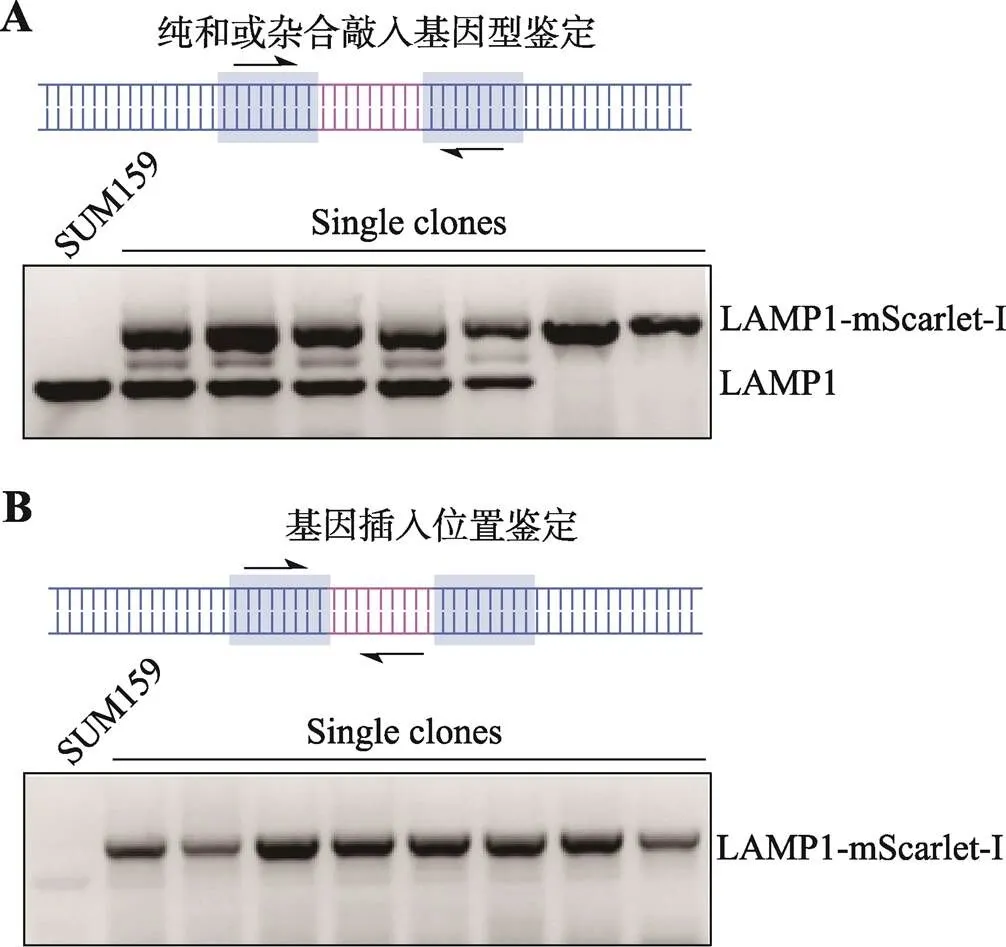
图5 利用PCR鉴定基因编辑表达LAMP1-mScarlet- I的单克隆细胞的基因型
A:利用PCR鉴定纯合和杂合单克隆细胞系的引物设计和琼脂糖凝胶电泳结果;B:利用PCR鉴定基因插入位点的引物设计和琼脂糖凝胶电泳结果。
5.4 单克隆细胞生长和形态观察
对于上述步骤鉴定出的阳性克隆,需要在普通光学显微镜下进行观察和鉴定。与野生型的细胞相比,选择细胞大小、形态、生长速度等相似的基因编辑细胞克隆并做好标记以备下一步鉴定,舍弃细胞形态等明显异常的基因编辑细胞克隆。
5.5 单克隆细胞蛋白表达鉴定
本步骤的目的是在蛋白水平上验证基因编辑的准确性以及检测基因编辑细胞系中目的蛋白的表达是否受到影响。通过BCA法测量每个样品的蛋白浓度并校准上样量后,采用Westen blot检测特定抗体结合的蛋白条带大小和条带的灰度值,进而判定基因编辑细胞系中目的蛋白是否与荧光蛋白标签融合表达,以及基因编辑后目的蛋白的表达水平是否发生变化。本文中mScarlet-I标记LAMP1的不同基因型的阳性克隆Westen blot鉴定结果如图6A。
此外,根据目的蛋白的不同,还可进行进一步的相应功能鉴定,以确保基因编辑或荧光标签的插入对目的蛋白的功能未造成影响。
5.6 单克隆细胞的成像鉴定和测序验证
将5.5步骤中挑选出的细胞克隆经胰酶消化后,将大部分细胞传代到6孔板进行培养以备冻存;另传代少许阳性细胞系克隆至成像皿上,16~24 h后在显微镜上进行荧光成像鉴定分析(图6B)。同时取少许细胞于PCR管中,进行第二次基因型和插入位点准确性的鉴定,操作流程同5.2和5.3。
6 结语
利用基因编辑技术,对内源目的蛋白进行快速准确的荧光标记,为研究蛋白质的定位、动力学和互作等提供了重要工具。例如,通过基因编辑技术技术对网格蛋白介导的内吞过程中的关键蛋白质分子进行荧光蛋白标记,结合活细胞单分子荧光成像,揭示了网格蛋白包被内吞囊泡组装、剪切和脱包被等过程中相关分子的动力学特征和机制[37,38]。本文对利用CRISPR-Cas9基因编辑技术,对目的蛋白进行荧光蛋白或自适应蛋白标签进行标记的实验设计和实验流程进行了梳理和介绍。基于本实验室在基因编辑细胞系构建中总结的经验,对细胞筛选富集和单克隆鉴定部分进行了详细介绍,以期为其他团队在哺乳动物细胞中进行基因编辑细胞系的构建提供参考,提高基因编辑细胞系构建的效率。本文介绍的CRISPR-Cas9基因编辑实验流程,也适用于构建如APEX2和Flag等标签敲入的细胞系。与此同时,多种其他的用于蛋白标签knock-in的基因编辑技术,例如利用Cas9D10A切口酶、CRISPR-Cpf1和非同源重组方法等[10,31,39],也展现出了在特定应用场景下的优势,研究者可根据研究的需要选取最合适的工具。
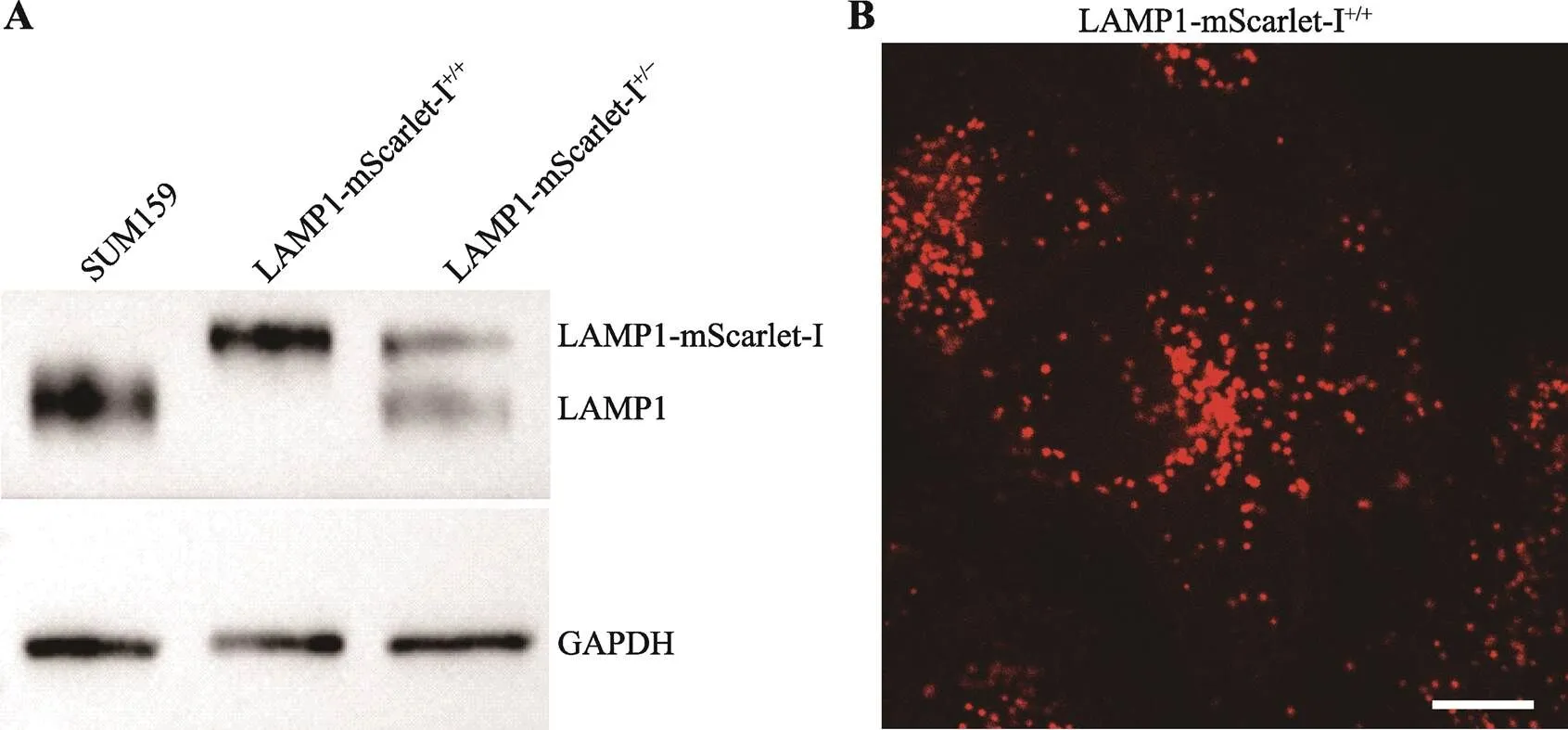
图6 Western blot和共聚焦成像鉴定基因编辑表达LAMP1-mScarlet-I的单克隆细胞
A:利用Western Blot鉴定纯合和杂合的单克隆细胞系;B:利用转盘式共聚焦显微镜对基因编辑表达LAMP1-mScarlet-I的单克隆细胞的中间层进行成像。标尺10 μm。
[1] Schneider AFL, Hackenberger CPR. Fluorescent labelling in living cells., 2017, 48: 61–68.
[2] Crivat G, Taraska JW. Imaging proteins inside cells with fluorescent tags., 2012, 30(1): 8–16.
[3] Cranfill PJ, Sell BR, Baird MA, Allen JR, Lavagnino Z, de Gruiter HM, Kremers GJ, Davidson MW, Ustione A, Piston DW. Quantitative assessment of fluorescent proteins., 2016, 13(7): 557–562.
[4] Hirano M, Ando R, Shimozono S, Sugiyama M, Takeda N, Kurokawa H, Deguchi R, Endo K, Haga K, Takai-Todaka R, Inaura S, Matsumura Y, Hama H, Okada Y, Fujiwara T, Morimoto T, Katayama K, Miyawaki A. A highly photostable and bright green fluorescent protein., 2022, 40(7): 1132–1142.
[5] Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV. HaloTag: a novel protein labeling technology for cell imaging and protein analysis., 2008, 3(6): 373–382.
[6] Hoelzel CA, Zhang X. Visualizing and manipulating biological processes by using HaloTag and SNAP-Tag technologies., 2020, 21(14): 1935–1946.
[7] Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system., 2013, 8(11): 2281–2308.
[8] Tamura R, Kamiyama D. CRISPR-Cas9-mediated knock- in approach to insert the GFP11 tag into the genome of a human cell line., 2023, 2564: 185–201.
[9] Surve S, Sorkin A. CRISPR/Cas9 gene editing of HeLa cells to tag proteins with mNeonGreen., 2022, 12(10): e4415.
[10] Koch B, Nijmeijer B, Kueblbeck M, Cai Y, Walther N, Ellenberg J. Generation and validation of homozygous fluorescent knock-in cells using CRISPR-Cas9 genome editing., 2018, 13(6): 1465–1487.
[11] Cabantous S, Terwilliger TC, Waldo GS. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein., 2005, 23(1): 102–107.
[12] Feng SY, Sekine S, Pessino V, Li H, Leonetti MD, Huang B. Improved split fluorescent proteins for endogenous protein labeling., 2017, 8(1): 370.
[13] Alford SC, Ding YD, Simmen T, Campbell RE. Dimerization-dependent green and yellow fluorescent proteins., 2012, 1(12): 569–575.
[14] Rodriguez EA, Campbell RE, Lin JY, Lin MZ, Miyawaki A, Palmer AE, Shu XK, Zhang J, Tsien RY. The growing and glowing toolbox of fluorescent and photoactive proteins., 2017, 42(2): 111–129.
[15] Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP)., 1996, 173(1 Spec No): 33–38.
[16] Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells., 2002, 296(5569): 913–916.
[17] Shaner NC, Lambert GG, Chammas A, Ni YH, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M, Davidson MW, Wang JW. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum., 2013, 10(5): 407–409.
[18] Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein., 2004, 22(12): 1567–1572.
[19] Merzlyak EM, Goedhart J, Shcherbo D, Bulina ME, Shcheglov AS, Fradkov AF, Gaintzeva A, Lukyanov KA, Lukyanov S, Gadella TWJ, Chudakov DM. Bright monomeric red fluorescent protein with an extended fluorescence lifetime., 2007, 4(7): 555–557.
[20] Bindels DS, Haarbosch L, van Weeren L, Postma M, Wiese KE, Mastop M, Aumonier S, Gotthard G, Royant A, Hink MA, Gadella TWJ. mScarlet: a bright monomeric red fluorescent protein for cellular imaging., 2017, 14(1): 53–56.
[21] Mukherjee S, Hung ST, Douglas N, Manna P, Thomas C, Ekrem A, Palmer AE, Jimenez R. Engineering of a brighter variant of the fusionRed fluorescent protein using lifetime Flow Cytometry and structure-guided mutations., 2020, 59(39): 3669–3682.
[22] Leonetti MD, Sekine S, Kamiyama D, Weissman JS, Huang B. A scalable strategy for high-throughput GFP tagging of endogenous human proteins., 2016, 113(25): E3501–E3508.
[23] Los GV, Wood K. The HaloTag: a novel technology for cell imaging and protein analysis., 2007, 356: 195–208.
[24] Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K. A general method for the covalent labeling of fusion proteins with small molecules in vivo., 2003, 21(1): 86–89.
[25] Juillerat A, Gronemeyer T, Keppler A, Gendreizig S, Pick H, Vogel H, Johnsson K. Directed evolution of O6- alkylguanine-DNA alkyltransferase for efficient labeling of fusion proteins with small molecules in vivo., 2003, 10(4): 313–317.
[26] Gautier A, Juillerat A, Heinis C, Corrêa IR, Kindermann M, Beaufils F, Johnsson K. An engineered protein tag for multiprotein labeling in living cells., 2008, 15(2): 128–136.
[27] Grimm JB, Brown TA, English BP, Lionnet T, Lavis LD. Synthesis of Janelia Fluor HaloTag and SNAP-Tag ligands and their use in cellular imaging experiments., 2017, 1663: 179–188.
[28] Trinh R, Gurbaxani B, Morrison SL, Seyfzadeh M. Optimization of codon pair use within the (GGGGS)3 linker sequence results in enhanced protein expression., 2004, 40(10): 717–722.
[29] van Rosmalen M, Krom M, Merkx M. Tuning the flexibility of glycine-serine linkers to allow rational design of multidomain proteins., 2017, 56(50): 6565– 6574.
[30] Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9- crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria., 2012, 109(39): E2579–E2586.
[31] Bukhari H, Müller T. Endogenous fluorescence tagging by CRISPR., 2019, 29(11): 912–928.
[32] Ratz M, Testa I, Hell SW, Jakobs S. CRISPR/Cas9- mediated endogenous protein tagging for RESOLFT super-resolution microscopy of living human cells., 2015, 5: 9592.
[33] Hendel A, Kildebeck EJ, Fine EJ, Clark J, Punjya N, Sebastiano V, Bao G, Porteus MH. Quantifying genome- editing outcomes at endogenous loci with SMRT sequencing., 2014, 7(1): 293–305.
[34] Paquet D, Kwart D, Chen A, Sproul A, Jacob S, Teo S, Olsen KM, Gregg A, Noggle S, Tessier-Lavigne M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9., 2016, 533(7601): 125–129.
[35] Concordet JP, Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens., 2018, 46(W1): W242– W245.
[36] Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, Virgin HW, Listgarten J, Root DE. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9., 2016, 34(2): 184–191.
[37] He KM, Marsland R, Upadhyayula S, Song E, Dang S, Capraro BR, Wang WM, Skillern W, Gaudin R, Ma MH, Kirchhausen T. Dynamics of phosphoinositide conversion in clathrin-mediated endocytic traffic., 2017, 552(7685): 410–414.
[38] He KM, Song E, Upadhyayula S, Dang S, Gaudin R, Skillern W, Bu K, Capraro BR, Rapoport I, Kusters I, Ma MH, Kirchhausen T. Dynamics of Auxilin 1 and GAK in clathrin-mediated traffic., 2020, 219(3): e201908142.
[39] Li GL, Yang SX, Wu ZF, Zhang XW. Recent developments in enhancing the efficiency of CRISPR/Cas9-mediated knock-in in animals., 2020, 42(7): 641–656.
李国玲, 杨善欣, 吴珍芳, 张献伟. 提高CRISPR/Cas9介导的动物基因组精确插入效率研究进展. 遗传, 2020, 42(7): 641–656.
The protocol of tagging endogenous proteins with fluorescent tags using CRISPR-Cas9 genome editing
Zhongsheng Wu1,2, Yu Gao1,2, Yongtao Du1,2, Song Dang1, Kangmin He1,2
The currently widely used CRISPR-Cas9 genome editing technology enables the editing of target genes (knock-out or knock-in) with high accuracy and efficiency. Guided by the small guide RNA, the Cas9 nuclease induces a DNA double-strand break at the targeted genomic locus. The DNA double-strand break can be repaired by the homology-directed repair pathway in the presence of a repair template. With the repair template containing the coding sequence of a fluorescent tag, the targeted gene can be inserted with the sequence of a fluorescent tag at the designed position. The genome editing mediated labeling of endogenous proteins with fluorescent tags avoids the potential artifacts caused by gene overexpression and substantially improves the reproductivity of imaging experiments. This protocol focuses on creating mammalian cell lines with endogenous proteins tagged with fluorescent proteins or self-labeling protein tags using CRISPR-Cas9 genome editing.
CRISPR-Cas9 genome editing; live cell; endogenous protein; fluorescent protein; self-labeling protein tag
2022-12-02;
2023-01-06;
2023-01-09
国家自然科学基金项目(编号:91957106,31970659)和国家重点研发计划(编号:2021YFA0804802,2022YFA1304500)资助[Supported by the National Natural Science Foundation of China (Nos. 91957106, 31970659), and the Ministry of Science and Technology of the People’s Republic of China (Nos. 2021YFA0804802, 2022YFA1304500)]
吴仲胜,博士研究生,专业方向:细胞脂质信号转导。E-mail: zswu@genetics.ac.cn
高誉,博士研究生,专业方向:细胞脂质信号转导。E-mail: gaoyu@genetics.ac.cn
杜勇涛,博士研究生,专业方向:细胞囊泡运输。E-mail: duyongtao@genetics.ac.cn
吴仲胜、高誉和杜勇涛并列第一作者。
何康敏,博士,研究员,研究方向:细胞囊泡运输和信号转导。E-mail: kmhe@genetics.ac.cn
10.16288/j.yczz.22-395
(责任编委: 史岸冰)
