基因渗入的检测和表征方法
2023-03-03刘泽璇赵毅强
刘泽璇,赵毅强
综 述
基因渗入的检测和表征方法
刘泽璇,赵毅强
中国农业大学生物学院,北京 100193
基因渗入也称为渗入性杂交,遗传学上指通过不断回交,遗传成分从一个群体的基因库流向另一个群体基因库的过程。基因渗入在自然界广泛存在,对于增加遗传多样性和提高环境适应性起到了重要的贡献,影响着动植物以及人类的演化进程。基因渗入作为进化中的重要事件而被广泛关注,包括鉴定基因渗入是否发生以及渗入的方向、时间和渗入模式等。随着高通量测序技术的快速发展,使得利用全基因组数据检测和表征基因渗入的方法不断出现。本文系统总结了基因渗入检测的系列方法,介绍了这些方法的设计原理和使用案例,并讨论了渗入后基因片段的维持与选择,以期为基因渗入相关研究提供较为全面的参考。
基因渗入;渗入检测;渗入方向;渗入时间;适应性渗入
基因渗入/渐渗(introgression)是发生在遗传距离相对较远的群体之间的基因流动,通常是经过种间杂交后由后代种与一个祖先种的反复回交所致[1]。作为一个长期的过程,基因渗入会导致复杂的、高度可变的基因组混合[2]。自Anderson[3]关于基因在物种间转移的开创性工作之后,渗入杂交(introgressive hybridization)一词开始进入研究者的视野,目前已在猪()、牛()、山羊()等动物、大量植物以及人类基因组中发现基因渗入的痕迹[4~7]。
通过基因渗入,一个群体的遗传物质转移至另一个群体中,进而对受体的基因库实现部分改写。过去20年中,越来越多的基因组数据表明基因渗入是引入新的遗传变异的重要方式[8],能丰富受体群体的变异类型,并增加遗传多样性。供体和受体之间的等位基因差异越大,供体方个体的数量越多,对受体群体遗传结构的影响就越大[9]。最著名的基因渗入的例子是尼安德特人对现代人祖先的基因渗入[4],研究表明非非洲人中约2%的基因组来自尼安德特人[10]。
基因渗入被认为是重要的进化力量。尽管远缘杂交可能导致杂交不育,然而如果后代两性可育,则有可能促使优势等位基因传递,最终产生进化上的优势[11,12]。来自供体的等位基因如果能提高受体群体所处环境下的适合度,此时的基因渗入就是适应性的。适应性基因渗入(adaptive introgression)通过引入新的适应性变异快速提高物种的适应能力[13]。适应性基因渗入在植物中进行了广泛的研究[14],据统计大约25%的开花植物与近亲交换基因,而动物中这一比例仅为10%[15]。由于杂交在动物界被认为是一种罕见的现象,因此基因渗入在动物进化中的作用一直被低估[13]。但随着高通量测序技术的普及,众多的古现代基因组数据及新发现的渗入事件为渗入在进化中的重要性提供了证据。近期的研究提示,从黑尾兔()中渗入的基因导致雪兔()产生多态性季节伪装,提高了环境适应性[16]。
基因渗入会在受体基因组中留下可检测的痕迹,相对于受体的遗传背景,渗入的遗传信息通常和供体更为相似[17]。近期的渗入会在子代个体基因组中保留清晰的印迹[18],然而多世代的回交将会显著减少保留印记的位点的数量,使得古老的基因渗入事件难以检测[19]。自Green等[4]利用全基因组数据检测现代人中尼安德特人的基因渗入以来,定性和定量检测基因渗入的方法快速发展。根据渗入后基因组特征的改变,检测策略包括遗传相似性、等位基因位点频率差异、拓扑结构改变等等。基于这些特征设计的统计量清晰直观,可解释性强。随着对基因渗入了解的加深,人们对是否存在渗入以及量化其在整个基因组中的程度、识别供体和受体群体、推断渗入事件发生时间等产生了浓厚的兴趣。虽然目前准确描述这些特征仍存在困难[20~22],但对还原渗入事件的全貌必不可少。本文对常用的基因渗入检测和表征方法进行了总结,并详细介绍了这些方法的设计原理及优缺点。
1 基因渗入的检测
基因渗入的检测主要包括判断渗入是否发生、具体位点及渗入比例。随着基因组数据海量增加,人们需要可靠的统计及计算方法检测群体历史中的渗入信号,目前已提出基于遗传相似性、位点频率差异等进行探究的统计量,其中统计量应用范围最广,并且进行了丰富的拓展[4,23]。
1.1 基因渗入检测统计量
1.1.1
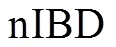
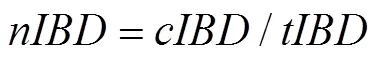
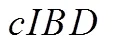
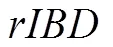
检测渗入的起点是个体之间的遗传相似性,可使用窗口为单位进行分析,主要用于基因渗入的定性分析。此方法侧重于个体层面的检测,敏感度较高,即使渗入时间短,渗入片段仅存在于少数个体中,也能够检测出来。检测还可以用于识别较大范围的基因渗入趋势,例如染色体间的差异[27],并且能够对候选区域进行定位,以便进一步表征。
1.1.2 卡方统计量
与个体间IBD的指标不同,单倍型相似性可以直接在群体水平进行比较。假设背景群体、受体群体为姐妹群体,那么二者的单倍型相似性较高。在不发生基因渗入的情况下,供体群体与受体群体整体的单倍型相似性较低。如果供体群体向受体群体发生基因渗入,则受体群体和供体群体在渗入区域整体的单倍型相似性会增加。卡方统计量常用于计数资料的相似性比较,此处用来衡量渗入区域的单倍型相似性,其公式为[28]:
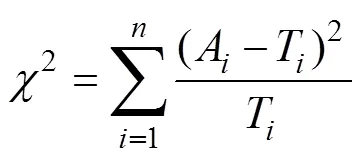
相比于单点的检测,单倍型包含了相位与连锁不平衡等丰富的信息,其组合数目和丰富程度大大增加了遗传信息的维度。与不同,卡方统计量直接基于群体水平进行检测,只有渗入在群体中达到一定程度时才会被检测出来;如果渗入发生在近期,或渗入单倍型只存在于少数个体中,则不会被鉴定出来。此外,由于该方法需要较多的样本量构建统计量,所以相应的成本较高。样本量不足时检测效力会严重降低[28]。
1.1.3统计量
统计量又被称为“ABBA-BABA”检验,通过计算不完全谱系分选(incomplete lineage sorting)的情况下两个姐妹物种与第三个密切相关的物种共享衍生等位基因的比例来推断基因渗入是否发生。统计量要求提供一个外群,计算公式如下:
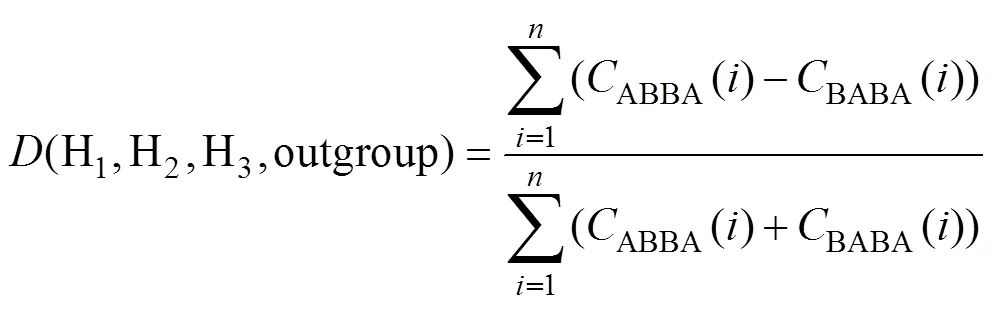
其中代表基因组上不同位置的等位基因位点;代表基因位点总数。H1与H2为两个姐妹群体, H3为渗入源群体,即供体群体。将外群等位基因定义为祖先等位基因A,衍生等位基因为B。假设外群祖先等位基因A是固定的,那么统计量就是ABBA和BABA模式之间的标准化差异。ABBA位点代表拓扑结构(((H2,H3),H1),O)下的模式,即H2和H3之间发生了基因渗入;BABA位点代表拓扑结构(((H1,H3),H2),O)下的模式,即H1和H3之间发生基因渗入。由于H1与H2起源于与H3分离的共同祖先,那么在随机交配的情况下出现在H3中的衍生等位基因应该和H1或者H2中的衍生等位基因频率相同。由此推断在没有基因渗入的情况下,统计量的期望值为0。当统计量为正时,表明H2群体与H3群体共享更多等位基因。相反地,当统计量为负值时则表明H1群体与H3群体共享更多等位基因。需要指出的是,统计量本身并不能给出渗入的方向。由于H3被预先定义为渗入供体,所以统计量为正时通常解释为H3到H2方向的渗入,为负时通常解释为H3到H1方向的渗入。
统计量首先用于检测现代人祖先和尼安德特人之间的基因渗入[4],现已广泛应用于各物种和群体的基因渗入检测。为了研究羊群之间的基因交流,Zheng等[5]设定H1为欧洲驯化山羊,H2为东亚驯化山羊,H3为阿塞拜疆和札格罗斯地区的野山羊,外群为盘羊。当H3为札格罗斯地区的野山羊时,检测到统计量为正值,表明该地区野山羊向东亚驯化山羊之间存在基因渗入;当H3为阿塞拜疆地区的野山羊时,检测到统计量为负值,表明该地区野山羊向欧洲驯化山羊之间存在基因渗入。在类似的研究中,Yu等[30]将H1设定为中国家猫(),H2为世界范围内家猫,H3为西藏山猫(),检测到山猫向同域家猫显著的渗入信号,为西藏山猫和同域家猫的基因交流提供了证据。
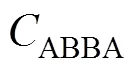
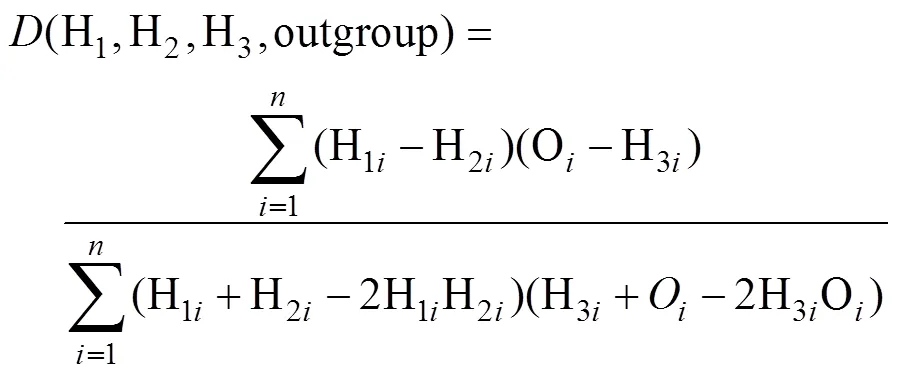
当用作非姐妹群体间渗入的全基因组测试时,在不同外群遗传距离、渗入方向、渗入模型等情况下统计量都具有稳健性[34,35]。统计量不适用于滑窗计算,因为较小的窗口中非重组基因座可能只存在单一位点模式,例如基因座只存在ABBA位点。此时值为1,会错误解读为群体H2和H3之间存在基因流。因此统计量更适合整个基因组范围的渗入检测,并且仅限于判断基因渗入是否发生,不能提供精确的信息。
1.1.44和4-ratio
-statistics由Pattreson等[32, 36]提出,用来衡量2个、3个或4个群体之间等位基因频率的差异。4个群体的-statistics与统计量非常相似,也广泛用于从群体遗传数据中检测基因交流,记为4-statistics(以下简称4),用来衡量群体H1、H2和H3、H4之间等位基因频率差异的平均相关性:
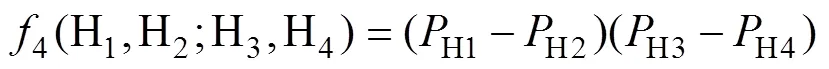
为等位基因频率,一般取多个SNP的均值。针对不同的拓扑结构,4存在以下代数变换:
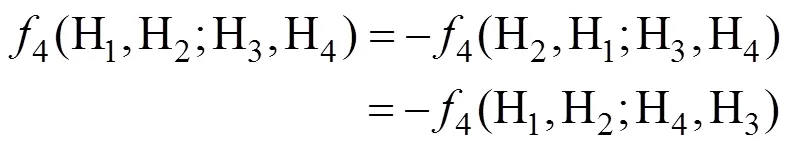
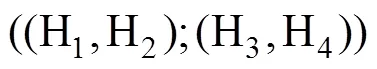
4-statistics另一个重要的应用是4-ratio,用于量化基因渗入的程度。假设图2中群体X由比例的H2群体和1–比例的H3群体混合而来,则有:
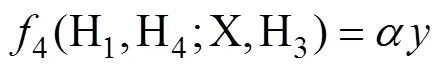
于是有:
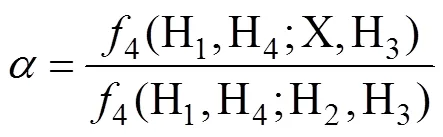
此处的即为4-ratio,代表来自群体H2的基因片段在群体X中比例。由于4-ratio计算简单,非常适合基因组规模的数据集并具有鲁棒性[31]。Green等[4]利用4-ratio,设定群体H1为黑猩猩(),H2为约鲁巴人,X为待测群体法国人,H3为供体尼安德特人,H4为丹尼索瓦人,通过4-ratio估计得到尼安德特人基因流入法国人群体的比例为0.022。
1.1.5 其他衍生统计量
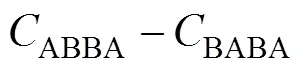
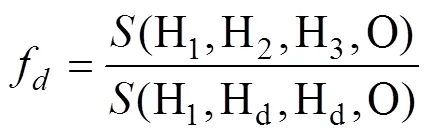
f的估计值与基因渗入水平大致成正比。在对蝴蝶(、、)[37]全基因组数据的分析中,通过多个不同群体组合测试f值,结果表明f统计量具有较好的稳健性。不足之处是估计值较保守,当群体数量很大时,f往往会低估渗入的真实水平,这是由于群体规模相对于分化时间较大时,谱系信息缺失无法准确量化渗入造成的[37]。
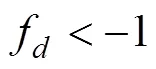
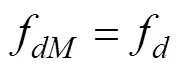
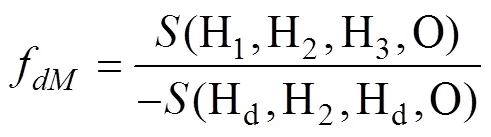
公式中Hd动态取H1或H3群体中具有更高衍生等位基因频率的群体。Vahedi等[39]设定H1为欧洲牛(Bos taurus),H2为复合肉牛,H3为印度牛(Bos taurus indicus),外群为牦牛(Bos grunniens),通过fdM统计显示约12.9%的复合肉牛群体的遗传成分由印度牛渗入。
A:4预期为0;B:4预期为;C:4预期为(1–)。4预期值由重叠路径确定。
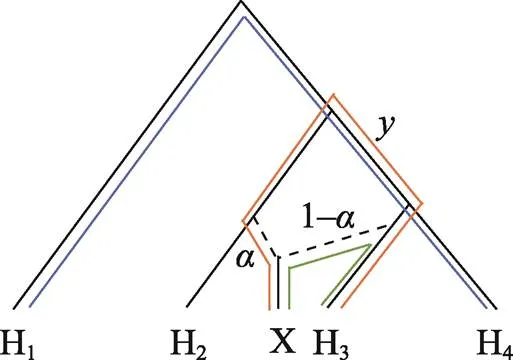
图2 f4-ratio五个群体下渗入图解
群体X祖先成分包括比例的H2群体和1–比例的H3群体。
f和f均适用于滑窗计算[23],能够准确计算渗入比例并具有良好的稳健性。
1.1.5.3D为了估算基因渗入的比例,Hamlin等[40]设计了统计量的扩展版本D。通过考虑ABBA和BABA以外的双等位基因位点模式,将BBAA位点的计数添加到分母中,此时分母包含所有双等位基因位点模式的计数,使得D接近于基因组的渗入比例。BBAA即位点基因与物种树拓扑结构(((H1,H2),H3),O)一致时的预期等位基因模式。
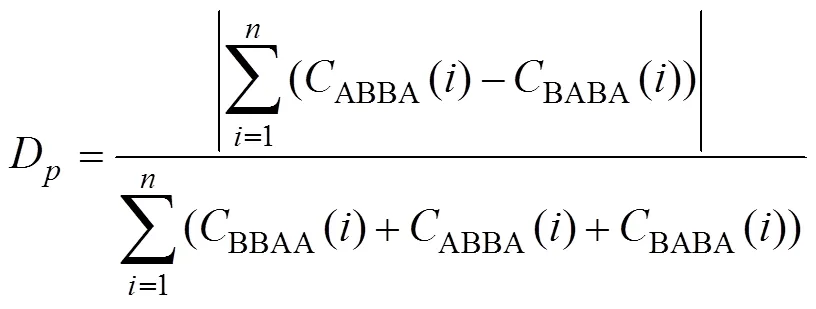
在不同渗入时间和渗入方向的模拟中,D估计值与基因渗入的真实值接近,呈线性变化,略微低估。尤其当基因渗入比例小于10%时,D与真实值差距小于1%[40]。Hamlin等[40]使用来自11个野生番茄()品种32个谱系的全基因组序列,通过D来评估密切相关种群间的渗入程度,以及地理因素、遗传差异等条件下渗入比例的变化。D既可以像f、f一样应用于滑窗计算,也可以用于量化全基因组的渗入比例。但是如果H1和H3之间以及H2和H3之间存在双向基因流,D可能会低估总的渗入量[40]。
以上统计量从不同角度对基因渗入进行定性或定量分析,总结见表1。由于这些统计量在设计的时候锚定基因渗入后某种基因组特征的改变,所以方法直观可解释性较强。统计量和statistics作为经典方法,是使用最多的全基因组渗入检测手段,同时在大多数群体统计情况下具有鲁棒性[31],适用于全局检测。f等拓展统计量针对渗入定量估计进行优化,矫正了和statistics的偏差,适用于滑窗计算。另外,渗入事件的其他参数,包括基因渗入的方向(供体和受体群体的身份区分)、渗入时间(相对于检测时间,渗入在多少代次之前发生)和渗入模式(离散的脉冲式或连续基因流)等对于准确了解渗入事件也同样重要。
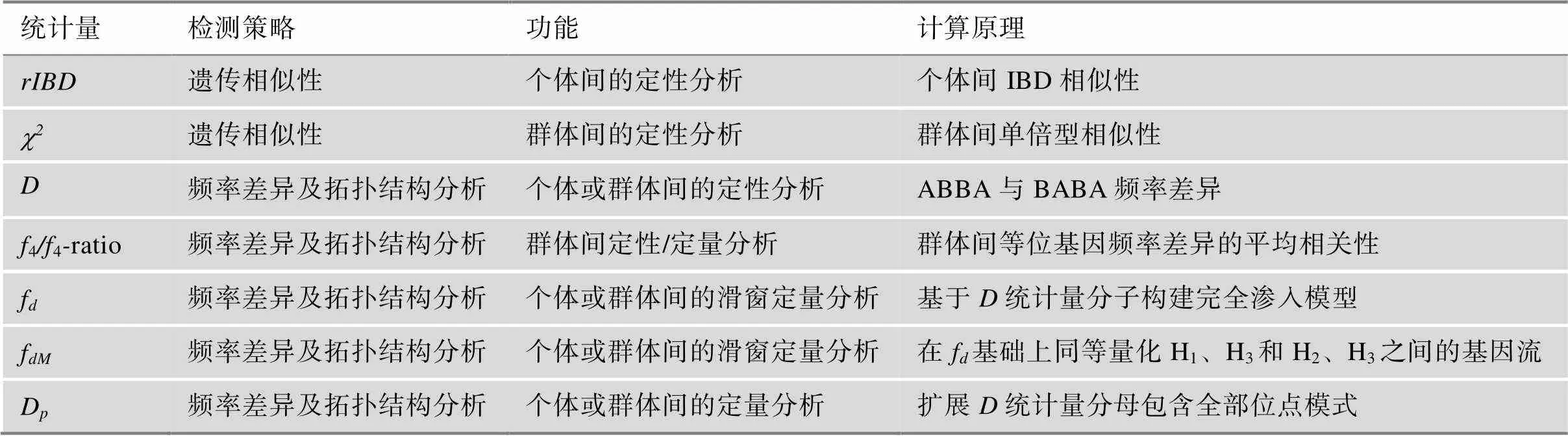
表1 渗入检测统计量汇总
1.2 基因渗入方向判定
虽然仅从基因树拓扑结构就可以了解到很多关于渗入的信息,但考虑基因树分支长度等额外信息可以提取更丰富的信息,例如确定群体之间的渗入方向,有助于精准描述渗入事件。本节主要介绍如何结合拓扑结构以外的信息推断渗入方向。
1.2.1 基于复杂拓扑结构
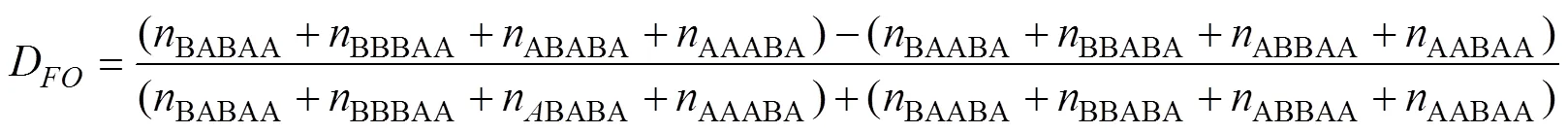
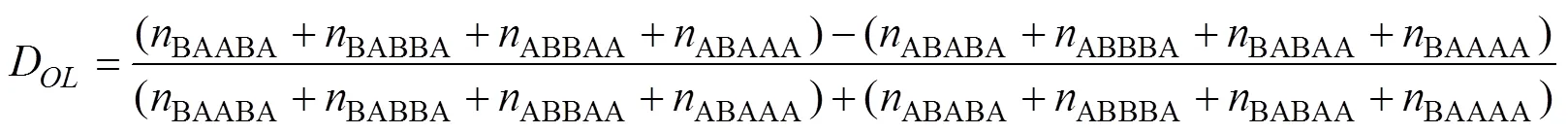
DFOIL的原理和统计量类似,都是比较不同渗入情况对应的拓扑结构下位点频率的差异,与预期值零的显著偏差表示渗入发生。不过该方法既考虑了待测群体之间的共享变异位点的情况,也考虑了待测群体之间保留共同祖先位点的情况。例如,D公式中包括BABAA和BBBAA(群体H1和群体H3共享变异位点)以及ABABA和AAABA(群体H1和群体H3保留共同祖先位点)。假设基因渗入方向为群体H2到群体H3,由于H1和H2为姐妹群体,H3将和H1共享更多的位点,D为正;D直接检测此类渗入,结果为正;由于发生渗入,H2和H3之间的共享等位基因比H1和H3之间多,D为负;最后,因为H1和H2都没有与H4发生渗入,H1和H4之间等位基因共享数目应与H2和H4之间的数目相同,D预计为零。不同渗入方向对应的各统计量值见表2。
数据模拟证明,DFOIL可以正确识别基因渗入发生的群体和渗入的方向,在一定的渗入量和渗入时间范围内准确推断出渗入事件,假阳性率极低[41]。Wang等[43]利用DFOIL,设H1、H2为欧洲和北美棕熊(),H3、H4为阿拉斯加北极熊(s)和古代北极熊,外群为黑熊,检测出古代和现代发生的从北极熊到棕熊的单向基因流。但是,DFOIL方法要求具有对称拓扑结构的4个群体,限制了方法的通用性。因为多个相同概率的差异拓扑结构是对称系统发育结构的特征[41],这也使得DFOIL无法应用于非对称的系统发育结构。例如非对称结构(((H1,H2),H3),H4),H1、H2与H3的关系比H4更密切,因此与H3共享更多等位基因,即使没有发生基因渗入D也会为正,导致该方法失去检测效力。
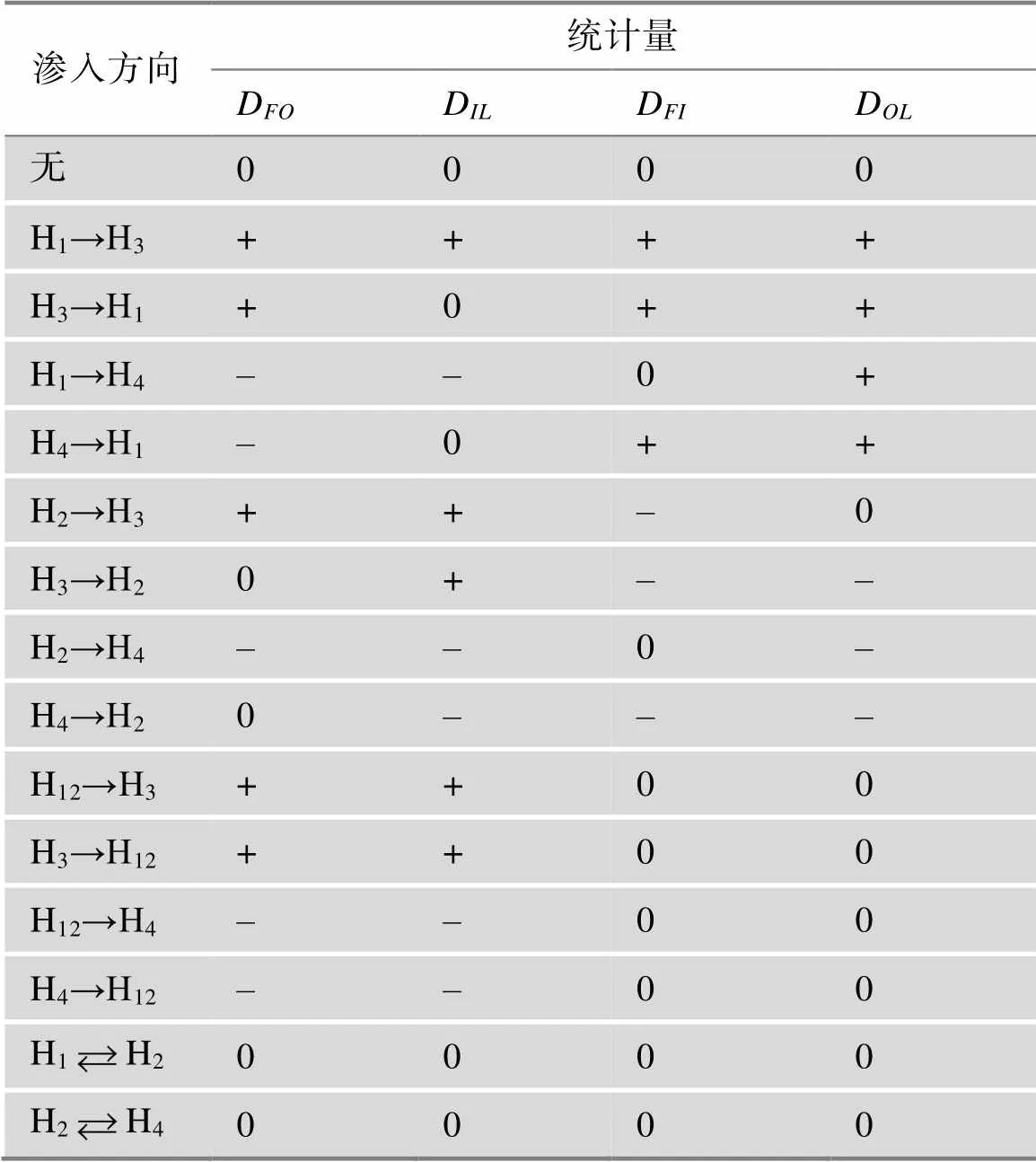
表2 预期渗入对应的DFOIL各统计量值
1.2.2 基于基因树分支长度
除了利用基因树拓扑结构,一些方法利用基因树分支长度差异来推断渗入方向。举例来说,下图中尽管H3到H2(图3B)和H2到H3(图3C)的渗入事件会产生相似的拓扑结构,但是这两个渗入事件产生的分支长度不同。群体H3到群体H2的基因流不会改变H1和H3的预期溯祖时间(图3B)。相反地,从群体H2到群体H3的基因流会使得群体H3中的位点可通过群体H2追溯其历史,同时也会使群体H1和群体H3更早地溯祖(图3C)。这种差异使人们能够以此确定不同群体之间主要的渗入方向。
基于此原理,2[44]统计量根据预期溯祖时间的差异进行推断,在特定基因树下区分基因渗入方向。假设群体H2和群体H3之间发生了基因渗入,此时2公式如下:
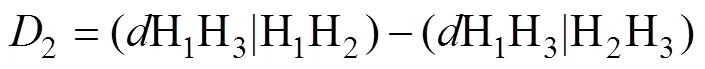
不同于DFOIL基于拓扑结构和等位基因频率差异,2在拓扑结构基础上考虑了基因树分支长度信息,仅需要3个待测群体和一个外群即可完成检测。模拟表明,如果群体分化时间较近,分支长度差异可能较小,2检测效力将大大下降,因此2无法检测近期发生的渗入事件。此外值得注意的是,2统计量提示的是基因渗入的主要方向,而不是排他性地说明基因渗入只在单方向发生。使用类似策略判断基因渗入方向的方法还有[45]统计量。
1.2.3 机器学习的应用
随着计算机技术的快速发展,机器学习方法用于推断基因渗入方向上也取得了一些进展。FILET[46]是一种有监督的机器学习方法,利用来自两个相关群体的基因组数据来确定给定的基因组窗口是否存在基因流,并判断渗入方向。此方法综合使用多个统计量,包括d[47]、G[48]以及作者为提高检测敏感性设计的d、d和Z[46]。
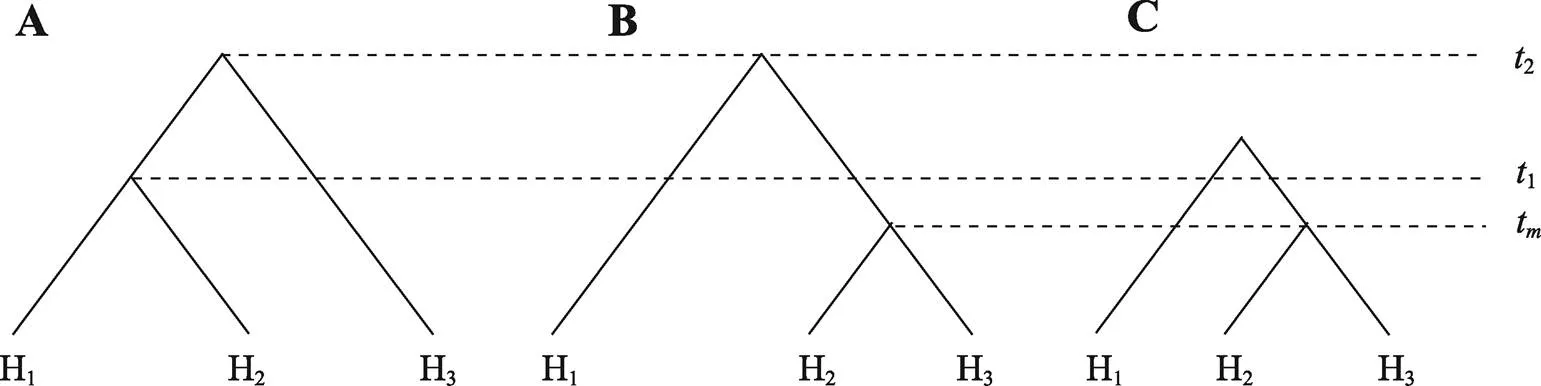
图3 不同渗入方向产生不同分支长度。
A:未发生基因交流的基因树;B:存在群体H3到群体H2基因流情况下的基因树;C:存在群体H2到群体H3基因流情况下的基因树。1表示H1和H2之间的溯祖时间,2表示H1和H2的共同祖先与谱系H3之间的溯祖时间,t表示H2和H3之间溯祖时间。
d代表了群体间最小的序列分歧度,G为d的拓展:
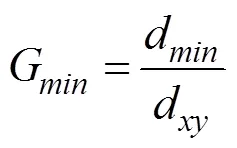
d统计量在群体A、B中比较了d和核苷酸多样性,分别为d、d、d,公式为:
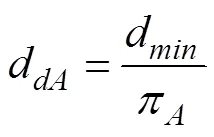
为群体A的核苷酸多样性。其原理为如果有群体A到群体B的渗入事件,并且群体B中至少有一条染色体保留了渗入的单倍型,则得到d值的两个个体应追溯到祖先群体A。因此,这两个个体之间该区域的平均序列分歧度应等于π。d与d类似。d为d值占所有比对的百分比排序。
Z基于在有基因流的情况下,受体群体中连锁不平衡(linkage disequilibrium,LD)会升高,但在全部群体中不会升高这一假设,其公式为:
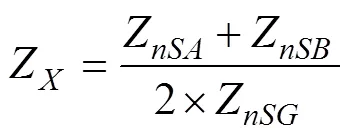
Z和Z分别测量群体A和群体B所有窗口的平均LD水平,Z测量全部群体内的LD水平。
FILET通过极端随机树分类器(extra-trees classifier),将基因组窗口分为群体A到群体B的基因渗入,群体B到群体A的基因渗入,以及没有渗入这3个类别。除了上述d、G、d、d和Z,此方法还使用了、[49]、[50]、F[51]和等统计量以提高渗入检测的敏感性。Schrider等[46]将FILET应用于果蝇数据,检测到果蝇(之间广泛的基因渗入,并推断出这种渗入主要是从到的方向进行的。
推断基因渗入的方向对于渗入细节的研究至关重要[52]。仅使用拓扑结构对应的位点频率无法区分渗入的方向,必须在检测过程中联合考虑其他信息。DFOIL通过增加群体数量,结合拓扑信息来推断所有假设方向,D基于基因树分支长度,无需多个群体也能够检测出渗入发生的主要方向。FILET则结合机器学习,通过引入多个统计量提高检测的敏感性,后续将问题框架转化为参数估计而非分类,或许能够精确推断渗入事件发生的时间和数量。
1.3 基因渗入时间估计
除了检测渗入事件是否发生、定量渗入比例、以及推断渗入方向,如果想要获得渗入历史的全貌,基因渗入在何时发生也同样受到研究者的关注。本节介绍利用基因组中重组信息和连锁不平衡水平来进行基因渗入时间的推断。
1.3.1 基于重组数
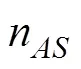
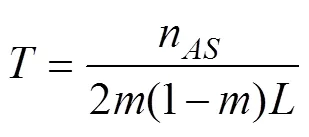
GLOBETROTTER[53]软件基于此原理估计渗入发生的时间。首先通过ChromoPainter[54]将待测群体的染色体拆分为单倍型区块,与其他群体的单倍型进行比对,寻找潜在的渗入供体。然后对每组渗入群体拟合一条共祖衰减曲线,衰减程度正比于渗入发生后的世代时间,从而估计出渗入发生时间。Hellenthal等[53]在考虑到渗入情况可能在多个种群间多次发生的情况下,通过GLOBETROTTER构建了全世界人类基因渗入历史的图谱,仔细区分了过去4000多年发生的100多个渗入事件。
在密切相关的群体之间,往往需要划分较大的单倍型区块以保证准确推断其祖先来源。这种情况下会导致n低估,相应的渗入时间也会被低估[55]。如果基因渗入程度较小,以至于检测不到渗入片段的重组事件,此方法则无法进行推断。因此应用前最好结合4ratio等统计量估计渗入比例,以判断基于重组数推断时间是否适用。
1.3.2 基于连锁不平衡
基因渗入初期会在受体群体中产生高水平的LD[20],由于每一代发生的重组导致特定等位基因彼此分离,子代群体中LD水平逐渐降低。根据基因组中LD衰减水平也可推断渗入发生的时间。
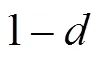
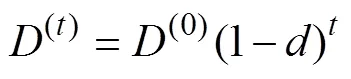
(0)代表初始的LD水平,
是渗入比例,是渗入发生的两个群体之间位点或的等位基因频率差异[57]。Moorjani等[58]首先基于此原理使用加权LD减少祖先LD水平的影响,进而来推断渗入时间[55]。
Patterson等[32]开发了ROLLOFF软件,基于图2的拓扑,模拟目标群体X中混合连锁不平衡的衰减,根据成对标记间的LD指数衰减率来估计单次渗入的发生时间:
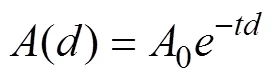
为成对标记间的遗传距离,0为初始LD水平,()为LD水平统计量,其结果与加权后的多态位点和LD片段有关,–td为经过代后成对标记间仍然处于LD的概率。模拟表明ROLLOFF能够准确推断500代以下渗入事件发生时间[32]。
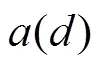
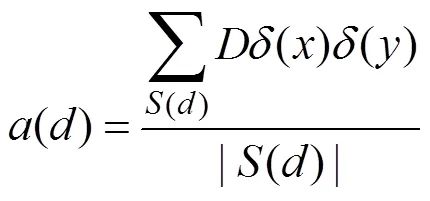
其中
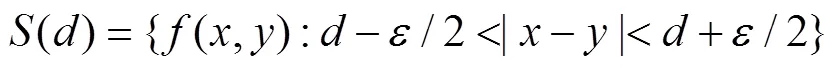
是的离散化参数,则有
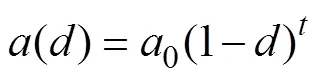
其中0是一个常数。根据待测群体LD水平拟合()的衰减曲线即可得出渗入发生的代次,并且此统计量对于参考群体的约束更少[59],相较于ROLLOFF,ALDER在单个群体祖先信息缺失时,仍然可以根据剩余的祖先群体(H2或H3)和受体群体(X)进行渗入时间推断。Yu等[30]通过ALDER计算的LD衰减程度,确定了从中国山猫到藏区同域家猫群体单向渗入的渗入时间约为7.42代之前。以家猫2年的世代时间计,即发生在大约15年前。
由于该方法不像ROLLOFF那样通过多态位点和LD片段进行标准化,因此该方法不会由于群体的样本经历了严重瓶颈效应或近期发生了基因渗入而产生偏差。此外ALDER通过快速傅里叶变换加速()计算,速度得到显著提升[33]。值得注意的是,ALDER必须校正背景LD水平,用来排除祖先群体中LD的干扰,使得加权LD曲线适用于待测群体距离较远的成对基因座[59]。
Pickrell等[60]将ALDER软件的检测范围扩展到群体经历多次基因渗入的情况,对于A、B两个群体以及A′、B′两个参考群体,
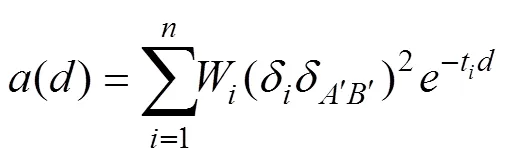
其中W代表渗入比例,是渗入发生的两个群体之间位点或的等位基因频率差异,代表参考群体中对应位点的等位基因频率差异,t为不同渗入事件的发生的代次。MALDER检测到的单次渗入事件与ALDER结果高度重合,同时会检测到少量的多次渗入。Busby等[61]利用MALDER检测撒哈拉以南非洲人群的渗入事件,发现来自相似祖先地区群体的渗入事件往往发生在同一时间并涉及相似的来源,表明共同的历史事件推动了该地区人群的群体结构变化。
经过数年的发展和改进,基于LD的渗入定年方法已十分稳健。对于年代较近(约35代内)的渗入事件估计较准确,同时随着样本量的增加估计的准确性增加[32,58,59]。与基于重组数的方法相比,此类方法可以基于基因型数据进行计算,因此更具有优势,能够降低局部祖先推断错误的风险。
2 基因渗入后的选择
目前大部分基因渗入的研究关注于渗入事件的定性和定量检测,对于渗入发生后基因组变化的报道相对较少。研究表明,由于遗传漂变或自然选择等多种因素的影响,渗入的遗传成分表现出动态的变化[9],并且随着时间推移,最终会由于漂变或选择固定下来。例如在向日葵()杂交品种中,基因组在不到1000代的时间内快速稳定[62]。尽管人们更加关注来自供体的适应性渗入,但基因组中的远源渗入部分仅有少量能保留下来[63]。自渗入以来2000代内,人类基因组中的尼安德特人片段少于4%被固定下来[64]。相比之下,在酵母菌()的杂交谱系中,近400代中约30%的基因组得到固定[65]。
2.1 有害渗入片段的清除
由于来自供体的基因与受体的基因组不相容[66]、生态不适应[67],以及供体本身携带有害等位基因[68]等原因,可能会造成杂交个体的适应性降低[69]。在这种情况下,基因组中渗入的部分会被选择性清除,并且在初期的数十代会经历一个快速净化期[70],净化速度主要与物种、重组率有关。前人的研究结果发现,选择更会倾向清除基因组中功能重要区域的渗入片段[63,64,68,69,71,72]。例如现代人中来自尼安德特人的渗入片段绝大部分被清除[72],在调控相关和保守的非编码区以及蛋白质编码序列的保守区域更是缺少尼安德特人来源的渗入片段[73]。与现代人不同,剑尾鱼()的渗入片段较大程度得以保留,但是在一些重要区域渗入片段也已被选择清除[63]。
重组在清除有害的基因渗入片段中发挥着重要的作用,研究表明重组率低的物种会更有效地清除渗入片段[9]。这是因为渗入的片段最初以完全连锁不平衡的方式出现在受体群体中,长的渗入片段可能聚合了有害效应,所以更容易被选择清除。随着渗入后代次的增加,重组将最初渗入的长片段打断为短片段并分散到不同的个体中。这些片段的危害较小,从而不易被选择清除[74]。
有效群体大小同样影响有害渗入片段的清除。有效群体小净化能力弱,小的受体群体更容易由于漂变的原因积累轻微的有害突变[9]。过量轻微有害突变的累积最终会降低个体的适合度并引发选择清除这些渗入片段,小的受体群体清除有害的基因渗入片段的速率更慢。与此相反,大群体则能更有效地清除渗入的有害变异。现代人的有效群体大小约为尼安德特人的10倍[75],较小的群体导致尼安德特人群体中逐渐积累大量的弱有害等位基因[76],渗入到现代人祖先群体中后,这些变异受到更强烈的选择作用[68],导致现代人中来自尼安德特人的祖先片段逐渐被剔除到低至约2%的水平[10]。
2.2 适应性渗入的保留
“适应性渗入”一词首次出现在杂交杜鹃()的研究中[77],之后随着越来越多渗入事件的发现,其在物种适应性和进化中的潜力受到关注,成为基因渗入研究的重点内容之一。来自供体的等位基因如果是因为提高了受体群体所处环境下的适合度而保留下来,即发生了适应性渗入。从本质上讲,这是一个选择性保留的过程,当适应性位点转向固定时,由于搭车效应,该区域周围杂合度降低。此外,受体群体和非渗入群体可能在适应性片段保留区域产生极端分化,而受体群体和供体群体间分化程度则较低[78]。由于适应性片段的选择性保留会和全基因组范围的选择清除同时发生[22],这在一定程度上变得容易区分,因为通常基因组中来自渗入供体的位点频率偏低,而适应性渗入片段在此背景下却具有较高的频率[72]。由此得出,不同选择力量共同塑造了渗入事件的结果[9],典型的例子是选择清除和适应性渗入在人类和尼安德特人的渗入事件中共同发挥作用[10, 79]。由于重组的缘故,适应性单倍型的长度并不会始终维持原始的长度,相反因为其留存的时间长,经历更多次数的重组,长度也只能维持在较短的水平[80]。
适应性渗入在进化中发挥着重要的作用,帮助群体快速适应环境并扩大生存范围[81],甚至促进物种进化乃至形成新的物种(表3)。适应性渗入最典型的例子之一是与高原适应性相关的基因渗入。Huerta-Sanchez等[82]通过对藏族人基因上下游区域重测序和单倍型分析,发现藏族人基因区域单倍型来自于丹尼索瓦人的基因渗入,帮助藏族人适应了高原缺氧的环境。作为高原适应性的明星基因,在高原犬中也发现了来自灰狼的基因渗入[83]。Wang等[84]进一步发现灰狼和喜马拉雅山狼群的基因的片段来自一种未知的古代犬科动物。Wang等[85]研究发现,牦牛贡献的基因有助于其他品种的牛适应高原低氧环境。
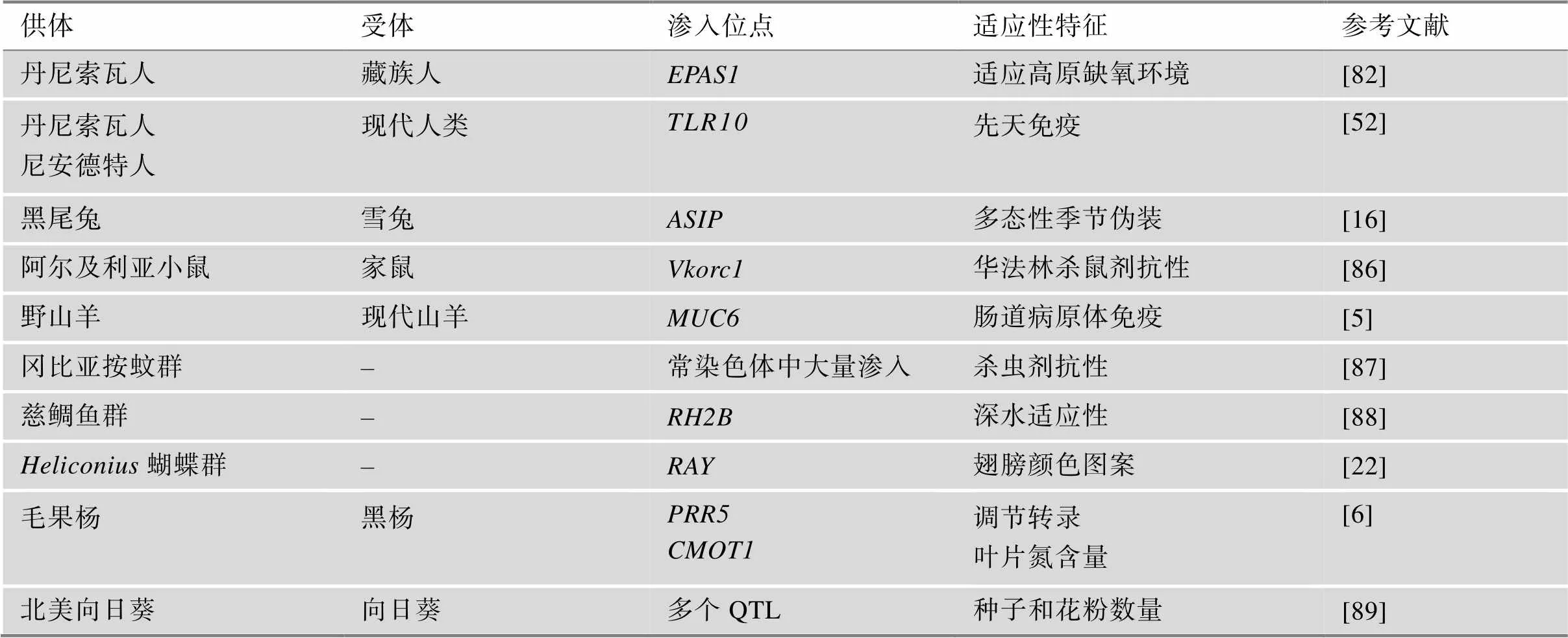
表3 影响广泛的适应性渗入汇总
3 结语与展望
本文介绍了检测基因渗入是否发生、判断渗入方向和估计渗入发生时间的方法和策略,并讨论了渗入发生后基因组结构的变化。由于基因渗入在学术和应用上的价值,使其在分子进化、物种保护以及相关领域越来越受到关注[20]。基因渗入在品种形成和性状获得方面起到了重要的作用。例如,有研究表明亚洲水稻的基因组中复杂的基因渗入帮助形成目前主要的粳稻和籼稻群体[90]。通过基因渗入,籼稻间接获得了主要的驯化等位基因,产生白色色素沉着、芳香和糯淀粉等特性。野山羊向现代山羊中渗入的基因有助于抵抗胃肠道病原体入侵[5]。作为一把双刃剑,Merotto等[91]发现水稻品种对咪唑啉酮除草剂的抗性基因渗入到杂草群体中,给农民造成了巨大的经济损失。基因渗入也可以作为遗传拯救的一种手段,通过引入外源适应性基因,减少因近亲繁殖而增加遗传负荷,提高群体的适应性从而避免灭绝[92]。
尽管与基因渗入相关的研究覆盖了对基因渗入事件定性和定量的检测及推断,但目前的研究对象更多是分化较远的物种或品种之间的基因交流。对于发生在分化距离较近的品种之间的渗入事件,当前广泛使用的以单位点频率变化为材料的检测方法效果欠佳,需要进一步改进统计分析方法,开发和优化基于单倍型等更高分辨率材料的检测方法和分析工具。另一方面,目前大多数推断渗入的方法基于已知的拓扑结构,基于没有基因渗入发生的溯祖模型来测试渗入假设。由于各种原因的限制,推测的溯祖模型并不能保证完全正确。拓扑结构的不确定性影响到渗入推断的可信度,如果选择错误的溯祖模型,那么渗入推断也可能会出现偏差。漫长的进化过程中可能存在已灭绝或未能采样到的幽灵谱系的渗入。幽灵谱系作为供体渗入到待测群体中,可能会对拓扑结构产生影响。假如幽灵谱系X渗入到群体H1中,群体拓扑结构可能从((H1,H2)H3)变为(H1(H2,H3))。此时H2、H3变为姐妹群体,表现出和H2、H3之间发生渗入相近的特征,从而导致误判。Tricou等[93]使用统计量进行测试,发现虽然幽灵谱系和待测群体之间能够检测出渗入发生,但是除此以外的信息推断都是错误的。此外,姐妹谱系间的渗入检测目前仍然较为困难。虽然姐妹谱系间的渗入在溯祖时间上会与其他渗入场景存在差异,但是这种信号很容易被连锁选择(linked selection)干扰[94],相关方法尚未被开发[19]。
在缺少先验知识的情况下,一些研究采取遍历的策略检测所有的拓扑组合,这样可能带来一些假阳性结果。如果更好地了解基因渗入供体和受体群体的遗传结构和品种历史,能对渗入事件有更清晰的把握。配合全基因组关联研究(genome wide association study,GWAS)更准确地绘制表型和适应性基因图谱[95,96],通过实验进一步验证基因渗入导致的适应性增强,将会更清楚地说明基因渗入是如何在进化中发挥作用。
感谢中国农业大学生物学院张春媛博士和王宇占博士对本文的指导。
[1] Rhymer JM, Simberloff D. Extinction by hybridization and introgression., 1996, 27: 83–109.
[2] Petit RJ, Excoffier L. Gene flow and species delimitation., 2009, 24(7): 386–393.
[3] Anderson E. Introgressive hybridization., 1953, 28(3): 280–307.
[4] Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai WW, Fritz MHY, Hansen NF, Durand EY, Malaspinas AS, Jensen JD, Marques-Bonet T, Alkan C, Prüfer K, Meyer M, Burbano HA, Good JM, Schultz R, Aximu-Petri A, Butthof A, Höber B, Höffner B, Siegemund M, Weihmann A, Nusbaum C, Lander ES, Russ C, Novod N, Affourtit J, Egholm M, Verna C, Rudan P, Brajkovic D, Kucan Ž, Gušic I, Doronichev VB, Golovanova LV, Lalueza-Fox C, de la Rasilla M, Fortea J, Rosas A, Schmitz RW, Johnson PLF, Eichler EE, Falush D, Birney E, Mullikin JC, Slatkin M, Nielsen R, Kelso J, Lachmann M, Reich D, Pääbo S. A draft sequence of the neandertal genome., 2010, 328(5979): 710–722.
[5] Zheng ZQ, Wang XH, Li M, Li YJ, Yang ZR, Wang XL, Pan XY, Gong M, Zhang Y, Guo YW, Wang Y, Liu J, Cai YD, Chen QM, Okpeku M, Colli L, Cai DW, Wang K, Huang SS, Sonstegard TS, Esmailizadeh A, Zhang WG, Zhang TT, Xu YB, Xu NY, Yang Y, Han JL, Chen L, Lesur J, Daly KG, Bradley DG, Heller R, Zhang GJ, Wang W, Chen YL, Jiang Y. The origin of domestication genes in goats., 2020, 6(21): eaaz5216.
[6] Suarez-Gonzalez A, Hefer CA, Christe C, Corea O, Lexer C, Cronk QCB, Douglas CJ. Genomic and functional approaches reveal a case of adaptive introgression from populus balsamifera (balsam poplar) in(black cottonwood)., 2016, 25(11): 2427–2442.
[7] Adavoudi R, Pilot M. Consequences of hybridization in mammals: a systematic review., 2021, 13(1): 50.
[8] Suarez-Gonzalez A, Lexer C, Cronk QCB. Adaptive introgression: a plant perspective., 2018, 14(3): 20170688.
[9] Moran BM, Payne C, Langdon Q, Powell DL, Brandvain Y, Schumer M. The genomic consequences of hybridization., 2021, 10: e69016.
[10] Prüfer K, De Filippo C, Grote S, Mafessoni F, Korlevic P, Hajdinjak M, Vernot B, Skov L, Hsieh PS, Peyrégne S, Reher D, Hopfe C, Nagel S, Maricic T, Fu QM, Theunert C, Rogers R, Skoglund P, Chintalapati M, Dannemann M, Nelson BJ, Key FM, Rudan P, Kućan Ž, Gušić I, Golovanova LV, Doronichev VB, Patterson N, Reich D, Eichler EE, Slatkin M, Schierup MH, Andrés AM, Kelso J, Meyer M, Pääbo S. A high-coverage neandertal genome from vindija cave in croatia., 2017, 358(6363): 655–658.
[11] Baack EJ, Rieseberg LH. A genomic view of introgression and hybrid speciation., 2007, 17(6): 513–518.
[12] Arnold ML. Transfer and origin of adaptations through natural hybridization: were anderson and stebbins right?, 2004, 16(3): 562–570.
[13] Taylor SA, Larson EL. Insights from genomes into the evolutionary importance and prevalence of hybridization in nature., 2019, 3(2): 170–177.
[14] Lexer C, Widmer A. The genic view of plant speciation: recent progress and emerging questions., 2008, 363(1506): 3023–3036.
[15] Mallet J, Besansky N, Hahn MW. How reticulated are species?, 2016, 38(2): 140–149.
[16] Jones MR, Mills LS, Alves PC, Callahan CM, Alves JM, Lafferty DJR, Jiggins FM, Jensen JD, Melo-Ferreira J, Good JM. Adaptive introgression underlies polymorphic seasonal camouflage in snowshoe hares., 2018, 360(6395): 1355–1358.
[17] Payseur BA, Rieseberg LH. A genomic perspective on hybridization and speciation., 2016, 25(11): 2337–2360.
[18] Gompert Z, Mandeville EG, Buerkle CA. Analysis of population genomic data from hybrid zones., 2017, 48(1): 207–229.
[19] Hibbins MS, Hahn MW. Phylogenomic approaches to detecting and characterizing introgression., 2022, 220(2): iyab173.
[20] Yuan K, Zhou Y, Ni XM, Wang YC, Liu C, Xu SH. Models, methods and tools for ancestry inference and admixture analysis., 2017, 5(3): 236–250.
[21] Jiao XY, Flouri T, Yang ZH. Multispecies coalescent and its applications to infer species phylogenies and cross- species gene flow., 2021, 8(12): nwab127.
[22] Edelman NB, Frandsen PB, Miyagi M, Clavijo B, Davey J, Dikow RB, Garcia-Accinelli G, Van Belleghem SM, Patterson N, Neafsey DE, Challis R, Kumar S, Moreira GRP, Salazar C, Chouteau M, Counterman BA, Papa R, Blaxter M, Reed RD, Dasmahapatra KK, Kronforst M, Joron M, Jiggins CD, Mcmillan WO, Di Palma F, Blumberg AJ, Wakeley J, Jaffe D, Mallet J. Genomic architecture and introgression shape a butterfly radiation., 2019, 366(6465): 594–599.
[23] Malinsky M, Matschiner M, Svardal H. Dsuite—fast D-statistics and related admixture evidence from VCF files., 2021, 21(2): 584–595.
[24] Bosse M, Megens HJ, Frantz LAF, Madsen O, Larson G, Paudel Y, Duijvesteijn N, Harlizius B, Hagemeijer Y, Crooijmans RPMA, Groenen MAM. Genomic analysis reveals selection for asian genes in european pigs following human-mediated introgression., 2014, 5: 4392.
[25] Giuffra E, Kijas JM, Amarger V, Carlborg O, Jeon JT, Andersson L. The origin of the domestic pig: independent domestication and subsequent introgression., 2000, 154(4): 1785–1791.
[26] Wang XT, Chen LY, Ma JX. Genomic introgression through interspecific hybridization counteracts genetic bottleneck during soybean domestication., 2019, 20(1): 22.
[27] Martin SH, Davey JW, Jiggins CD. Evaluating the use of ABBA-BABAa statistics to locate introgressed loci., 2015, 32(1): 244–257.
[28] Zhang CY, Lin D, Wang YZ, Peng DZ, Li HF, Fei J, Chen KW, Yang N, Hu XX, Zhao YQ, Li N. Widespread introgression in chinese indigenous chicken breeds from commercial broiler., 2019, 12(3): 610–621.
[29] Wang YZ, Zhang CY, Peng YB, Cai XY, Hu XX, Bosse M, Zhao YQ. Whole-genome analysis reveals the hybrid formation of chinese indigenous DHB pig following human migration., 2022, 15(3): 501–514.
[30] Yu H, Xing YT, Meng H, He B, Li WJ, Qi XZ, Zhao JY, Zhuang Y, Xu X, Yamaguchi N, Driscoll CA, O'brien SJ, Luo SJ. Genomic evidence for the chinese mountain cat as a wildcat conspecific () and its introgression to domestic cats., 2021, 7(26): eabg0221.
[31] Durand EY, Patterson N, Reich D, Slatkin M. Testing for ancient admixture between closely related populations., 2011, 28(8): 2239–2252.
[32] Patterson N, Moorjani P, Luo YT, Mallick S, Rohland N, Zhan YP, Genschoreck T, Webster T, Reich D. Ancient admixture in human history., 2012, 192(3): 1065–1093.
[33] Harris AM, Degiorgio M. Admixture and ancestry inference from ancient and modern samples through measures of population genetic drift., 2017, 89(1): 21–46.
[34] Zheng YC, Janke A. Gene flow analysis method, the D-statistic, is robust in a wide parameter space., 2018, 19(1): 10.
[35] Kong S, Kubatko LS. Comparative performance of popular methods for hybrid detection using genomic data., 2021, 70(5): 891–907.
[36] Reich D, Thangaraj K, Patterson N, Price AL, Singh L. Reconstructing indian population history., 2009, 461(7263): 489–494.
[37] Martin SH, Davey JW, Salazar C, Jiggins CD. Recombination rate variation shapes barriers to introgression across butterfly genomes., 2019, 17(2): e2006288.
[38] Malinsky M, Challis RJ, Tyers AM, Schiffels S, Terai Y, Ngatunga BP, Miska EA, Durbin R, Genner MJ, Turner GF. Genomic islands of speciation separate cichlid ecomorphs in an east african crater lake., 2015, 350(6267): 1493–1498.
[39] Vahedi SM, Ardestani SS, Afshari KP, Ghoreishifar SM, Moghaddaszadeh-Ahrabi S, Banabazi MH, Brito LF. Genome-wide selection signatures and human-mediated introgression events inindicus-influenced composite beef cattle., 2022, 13: 844653.
[40] Hamlin JAP, Hibbins MS, Moyle LC. Assessing biological factors affecting postspeciation introgression., 2020, 4(2): 137–154.
[41] Pease JB, Hahn MW. Detection and polarization of introgression in a five-taxon phylogeny., 2015, 64(4): 651–662.
[42] Martin SH, Amos W. Signatures of introgression across the allele frequency spectrum., 2021, 38(2): 716–726.
[43] Wang MS, Murray GGR, Mann D, Groves P, Vershinina AO, Supple MA, Kapp JD, Corbett-Detig R, Crump SE, Stirling I, Laidre KL, Kunz M, Dalén L, Green RE, Shapiro B. A polar bear paleogenome reveals extensive ancient gene flow from polar bears into brown bears., 2022, 6(7): 936–944.
[44] Hibbins MS, Hahn MW. The timing and direction of introgression under the multispecies network coalescent., 2019, 211(3): 1059–1073.
[45] Forsythe ES, Sloan DB, Beilstein MA. Divergence-based introgression polarization., 2020, 12(4): 463–478.
[46] Schrider DR, Ayroles J, Matute DR, Kern AD. Supervised machine learning reveals introgressed loci in the genomes of drosophila simulans and., 2018, 14(4): e1007341.
[47] Joly S, Mclenachan PA, Lockhart PJ. A statistical approach for distinguishing hybridization and incomplete lineage sorting., 2009, 174(2): E54–E70.
[48] Geneva AJ, Muirhead CA, Kingan SB, Garrigan D. A new method to scan genomes for introgression in a secondary contact model., 2015, 10(4): e0118621.
[49] Hudson RR. A new statistic for detecting genetic differentiation., 2000, 155(4): 2011–2014.
[50] Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism., 1989, 123(3): 585–595.
[51] Hudson RR, Slatkin M, Maddison WP. Estimation of levels of gene flow from DNA sequence data., 1992, 132(2): 583–589.
[52] Dannemann M, Andrés AM, Kelso J. Introgression of neandertal- and denisovan-like haplotypes contributes to adaptive variation in human toll-like receptors., 2016, 98(1): 22–33.
[53] Hellenthal G, Busby GBJ, Band G, Wilson JF, Capelli C, Falush D, Myers S. A genetic atlas of human admixture history., 2014, 343(6172): 747–751.
[54] Lawson DJ, Hellenthal G, Myers S, Falush D. Inference of population structure using dense haplotype data., 2012, 8(1): e1002453.
[55] Chimusa ER, Defo J, Thami PK, Awany D, Mulisa DD, Allali I, Ghazal H, Moussa A, Mazandu GK. Dating admixture events is unsolved problem in multi-way admixed populations., 2020, 21(1): 144–155.
[56] Hill WG, Robertson A. The effect of linkage on limits to artificial selection., 1966, 8(3): 269–294.
[57] Winkler CA, Nelson GW, Smith MW. Admixture mapping comes of age., 2010, 11: 65–89.
[58] Moorjani P, Patterson N, Hirschhorn JN, Keinan A, Hao L, Atzmon G, Burns E, Ostrer H, Price AL, Reich D. The history of african gene flow into southern europeans, levantines, and jews., 2011, 7(4): e1001373.
[59] Loh PR, Lipson M, Patterson N, Moorjani P, Pickrell JK, Reich D, Berger B. Inferring admixture histories of human populations using linkage disequilibrium., 2013, 193(4): 1233–1254.
[60] Pickrell JK, Patterson N, Loh PR, Lipson M, Berger B, Stoneking M, Pakendorf B, Reich D. Ancient west eurasian ancestry in southern and eastern africa., 2014, 111(7): 2632–2637.
[61] Busby GB, Band G, Le QS, Jallow M, Bougama E, Mangano VD, Amenga-Etego LN, Enimil A, Apinjoh T, Ndila CM, Manjurano A, Nyirongo V, Doumba O, Rockett KA, Kwiatkowski DP, Spencer CC, Malaria Genomic Epidemiology Network. Admixture into and within sub-saharan africa., 2016, 5: e15266.
[62] Buerkle CA, Rieseberg LH. The rate of genome stabilization in homoploid hybrid species., 2008, 62(2): 266–275.
[63] Schumer M, Cui RF, Powell DL, Rosenthal GG, Andolfatto P. Ancient hybridization and genomic stabilization in a swordtail fish., 2016, 25(11): 2661–2679.
[64] Sankararaman S, Mallick S, Dannemann M, Prüfer K, Kelso J, Pääbo S, Patterson N, Reich D. The genomic landscape of neanderthal ancestry in present-day humans., 2014, 507(7492): 354–357.
[65] Stukenbrock EH, Christiansen FB, Hansen TT, Dutheil JY, Schierup MH. Fusion of two divergent fungal individuals led to the recent emergence of a unique widespread pathogen species., 2012, 109(27): 10954–10959.
[66] Orr HA. The population genetics of speciation: the evolution of hybrid incompatibilities., 1995, 139(4): 1805–1813.
[67] Schluter D. Evidence for ecological speciation and its alternative., 2009, 323(5915): 737–741.
[68] Harris K, Nielsen R. The genetic cost of neanderthal introgression., 2016, 203(2): 881–891.
[69] Sankararaman S, Mallick S, Patterson N, Reich D. The combined landscape of denisovan and neanderthal ancestry in present-day humans., 2016, 26(9): 1241–1247.
[70] Veller C, Edelman NB, Muralidhar P, Nowak MA. Recombination and selection against introgressed DNA., 2021.
[71] Kulmuni J, Nouhaud P, Pluckrose L, Satokangas I, Dhaygude K, Butlin RK. Instability of natural selection at candidate barrier loci underlying speciation in wood ants., 2020, 29(20): 3988–3999.
[72] Vernot B, Akey JM. Resurrecting surviving neandertal lineages from modern human genomes., 2014, 343(6174): 1017–1021.
[73] Petr M, Pääbo S, Kelso J, Vernot B. Limits of long-term selection against neandertal introgression., 2019, 116(5): 1639–1644.
[74] Barton NH, Otto SP. Evolution of recombination due to random drift., 2005, 169(4): 2353–2370.
[75] Juric I, Aeschbacher S, Coop G. The strength of selection against neanderthal introgression., 2016, 12(11): e1006340.
[76] Prüfer K, Racimo F, Patterson N, Jay F, Sankararaman S, Sawyer S, Heinze A, Renaud G, Sudmant PH, De Filippo C, Li H, Mallick S, Dannemann M, Fu QM, Kircher M, Kuhlwilm M, Lachmann M, Meyer M, Ongyerth M, Siebauer M, Theunert C, Tandon A, Moorjani P, Pickrell J, Mullikin JC, Vohr SH, Green RE, Hellmann I, Johnson PLF, Blanche H, Cann H, Kitzman JO, Shendure J, Eichler EE, Lein ES, Bakken TE, Golovanova LV, Doronichev VB, Shunkov MV, Derevianko AP, Viola B, Slatkin M, Reich D, Kelso J, Pääbo S. The complete genome sequence of a neanderthal from the altai mountains., 2014, 505(7481): 43–49.
[77] Milne RI, Abbott RJ. Origin and evolution of invasive naturalized material of. In the british isles., 2000, 9(5): 541–556.
[78] Ai HS, Fang XD, Yang B, Huang ZY, Chen H, Mao LK, Zhang F, Zhang L, Cui LL, He WM, Yang J, Yao XM, Zhou LS, Han LJ, Li J, Sun SL, Xie XH, Lai BX, Su Y, Lu Y, Yang H, Huang T, Deng WJ, Nielsen R, Ren J, Huang LS. Adaptation and possible ancient interspecies introgression in pigs identified by whole-genome sequencing., 2015, 47(3): 217–225.
[79] Zeberg H, Pääbo S. The major genetic risk factor for severe COVID-19 is inherited from neanderthals., 2020, 587(7835): 610–612.
[80] Shchur V, Svedberg J, Medina P, Corbett-Detig R, Nielsen R. On the distribution of tract lengths during adaptive introgression., 2020, 10(10): 3663–3673.
[81] Edelman NB, Mallet J. Prevalence and adaptive impact of introgression., 2021, 55: 265–283.
[82] Huerta-Sánchez E, Jin X, Asan, Bianba Z, Peter BM, Vinckenbosch N, Liang Y, Yi X, He MZ, Somel M, Ni PX, Wang B, Ou XH, Huasang, Luosang JB, Cuo ZXP, Li K, Gao GY, Yin Y, Wang W, Zhang XQ, Xu X, Yang HM, Li YR, Wang J, Wang J, Nielsen R. Altitude adaptation in tibetans caused by introgression of denisovan-like DNA., 2014, 512(7513): 194–197.
[83] Vonholdt B, Fan ZX, Vecchyo DOD, Wayne RK.variants in high altitude tibetan wolves were selectively introgressed into highland dogs., 2017, 5: e3522.
[84] Wang MS, Wang S, Li Y, Jhala Y, Thakur M, Otecko NO, Si JF, Chen HM, Shapiro B, Nielsen R, Zhang YP, Wu DD. Ancient hybridization with an unknown population facilitated high-altitude adaptation of canids., 2020, 37(9): 2616–2629.
[85] Wang XG, Ju ZH, Jiang Q, Zhong JF, Liu CK, Wang JP, Hoff JL, Schnabel RD, Zhao H, Gao YP, Liu WH, Wang LL, Gao YD, Yang CH, Hou MH, Huang N, Regitano LCA, Porto-Neto LR, Decker JE, Taylor JF, Huang JM. Introgression, admixture, and selection facilitate genetic adaptation to high-altitude environments in cattle., 2021, 113(3): 1491–1503.
[86] Liu KJ, Steinberg E, Yozzo A, Song Y, Kohn MH, Nakhleh L. Interspecific introgressive origin of genomic diversity in the house mouse., 2015, 112(1): 196–201.
[87] Fontaine MC, Pease JB, Steele A, Waterhouse RM, Neafsey DE, Sharakhov IV, Jiang XF, Hall AB, Catteruccia F, Kakani E, Mitchell SN, Wu YC, Smith HA, Love RR, Lawniczak MK, Slotman MA, Emrich SJ, Hahn MW, Besansky NJ. Extensive introgression in a malaria vector species complex revealed by phylogenomics., 2015, 347(6217): 1258524.
[88] Malinsky M, Svardal H, Tyers AM, Miska EA, Genner MJ, Turner GF, Durbin R. Whole-genome sequences ofreveal multiple radiations interconnected by gene flow., 2018, 2(12): 1940–1955.
[89] Whitney KD, Broman KW, Kane NC, Hovick SM, Randell RA, Rieseberg LH. Quantitative trait locus mapping identifies candidate alleles involved in adaptive introgression and range expansion in a wild sunflower., 2015, 24(9): 2194–2211.
[90] Choi JY, Platts AE, Fuller DQ, Hsing YI, Wing RA, Purugganan MD. The rice paradox: multiple origins but single domestication in asian rice., 2017, 34(4): 969–979.
[91] Merotto A, Goulart ICGR, Nunes AL, Kalsing A, Markus C, Menezes VG, Wander AE. Evolutionary and social consequences of introgression of nontransgenic herbicide resistance from rice to weedy rice in brazil., 2016, 9(7): 837–846.
[92] Hoffmann AA, Miller AD, Weeks AR. Genetic mixing for population management: from genetic rescue to provenancing., 2020, 14(3): 634–652.
[93] Tricou T, Tannier E, De Vienne DM. Ghost lineages highly influence the interpretation of introgression tests., 2022, 71(5): 1147–1158.
[94] Roux C, Fraïsse C, Romiguier J, Anciaux Y, Galtier N, Bierne N. Shedding light on the grey zone of speciation along a continuum of genomic divergence., 2016, 14(12): e2000234.
[95] Zhou X, Carbonetto P, Stephens M. Polygenic modeling with bayesian sparse linear mixed models., 2013, 9(2): e1003264.
[96] Gompert Z, Egan SP, Barrett RDH, Feder JL, Nosil P. Multilocus approaches for the measurement of selection on correlated genetic loci., 2017, 26(1): 365–382.
Methods to detect and characterize introgression
Zexuan Liu, Yiqiang Zhao
Introgression, also known as introgressive hybridization, refers to the process that genetic components from the gene pool of one population transfer to the other via constant backcrossing. Introgression is widespread in nature, which plays important roles in increasing genetic diversity and improving adaptability to the environment, and in turn, influences the evolutionary progress of animals, plants and humans. Being as an important evolutionary event, researchers pay great attention to the detection of introgression, the introgression direction, the introgression timing, the pattern of introgression and so on. With the rapid development of high-throughput sequencing technologies, methods to detect and characterize introgression based on genome-wide data are continuously developed. In this review, we summarize a series of methods for introgression detection, and introduce the design principles and applications of these methods. We also discuss the maintenance and selection of gene segments after introgression. This review provides a relatively comprehensive reference for the studies on introgression.
introgression; introgression detection; introgression direction; introgression timing; adaptive introgression
2022-12-01;
2023-01-04;
2023-01-26
国家重点研发计划项目(编号:2021YFD1200803)资助[Supported by the National Key Research and Development Program of China (No. 2021YFD1200803)]
刘泽璇,在读博士研究生,专业方向:生物信息学。E-mail: liuzexuan@cau.edu.cn
赵毅强,博士,副教授,博士生导师,研究方向:生物信息学。E-mail: yiqiangz@cau.edu.cn
10.16288/j.yczz.22-394
(责任编委: 姜雨)
