基于BSA-seq技术对豌豆花色基因的精细定位
2023-02-27李孟伟李正丽宗绪晓
严 昕 项 超 刘 荣 李 冠 李孟伟 李正丽 宗绪晓,* 杨 涛,*
基于BSA-seq技术对豌豆花色基因的精细定位
严 昕1,**项 超2,**刘 荣1李 冠1李孟伟1李正丽3宗绪晓1,*杨 涛1,*
1中国农业科学院作物科学研究所, 北京 100081;2四川省农业科学院作物科学所, 四川成都 610066;3贵州省农业科学院园艺研究所, 贵州贵阳 550006
BSA-seq技术在挖掘农艺性状相关的新基因中已被广泛应用, 随着豌豆首个参考基因组问世, 将BSA-seq技术结合豌豆基因组的基因定位策略势在必行。本研究利用紫花亲本G0004562、白花亲本G0002930以及F2群体, 通过BSA-seq技术对豌豆花色基因进行初步定位, 获得31.42 Mb定位区间, 再通过设计InDel分子标记分析进一步缩小定位区间, 最终将目标基因定位在包含19个基因的0.99 Mb区间内, 通过基因注释信息推测出为豌豆花色候选基因。本研究结果验证了BSA-seq技术快速高效定位豌豆花色基因的可行性, 为利用该技术挖掘豌豆其他重要农艺性状相关基因奠定了基础。
豌豆; BSA-seq; InDel; 基因定位
豌豆(L., 2=2=14)是我国第一大食用豆类作物[1], 在我国的主要产区是西南地区、长三角地区和两湖地区。由于其蛋白质含量高、可获得性好、生产成本低, 豌豆已经成为食品热门领域植物基蛋白开发中最受欢迎的原料之一。据FAO最新统计, 全球鲜豌豆的栽培面积、总产量分别占全球的55.32%和56.63%[2]。同时豌豆具有生物固氮作用, 是我国调整种植业结构的重要经济作物[3]。
目前豌豆中大部分基因定位都是通过同源克隆或基于构建遗传图谱的QTL定位获得。同源克隆利用基因在进化过程中的保守性实现基因的精细定位,未完成测序或缺少转录信息的物种可以利用基因的保守区域通过基因信息较多的物种研究或克隆该基因, 如豌豆的抗白粉病的基因[4]控制开花信号的基因[5], 调控开花和花序发育的基因[6]。利用模式植物蒺藜苜蓿的参考基因组, 可以为豌豆后续精细作图、功能分析以及基因克隆等提供参考信息, 但是进行基因图位克隆会存在一定的风险性和不准确性, 并且豌豆基因组中大约有54%的序列在蒺藜苜蓿基因组内无法找到同源的片段。QTL定位是通过分子标记与数量性状表型值之间的关系, 检测数量性状位点的存在, 并利用多种作图方法绘制遗传连锁图谱[7-9], 确定某一数量性状的数目、位置、遗传效应等, 如Wu等[10]利用13.2 K SNP阵列和SSR标记通过QTL作图确定了8个与豌豆根腐病抗性相关的主效QTL, Aznar-Fernández等[11]整合出平均标记密度为0.38 cM遗传连锁图谱, 确定了与豌豆象抗性相关的3个QTL。QTL定位优点是不需要完成物种参考基因组测序也能将基因定位在连锁群上, 通常构建一个群体可以同时实现多个主效或微效数量性状基因的定位。但是QTL定位的准确性容易受环境以及开发的标记是否具有多态性等影响, 同时RIL群体和NIL群体作为QTL定位的首选材料, 其构建群体又需要的时间相对较长, 整个过程费时费力, 是一项劳动密集且成本较高的工作。另外, 对于QTL的精细定位, 须开发另一个数千系的大型群体, 在目标区域产生足够的重组体, 以进行进一步的精细定位。
近年来随着越来越多的物种完成了全基因组测序, 以及高通量测序技术的迭代与发展, 推进了BSA-seq技术的应用, 先后衍生出了Mutmap、QTL- seq、RAD-seq和SLAF-seq技术。BSA-seq的应用从模式物种拓展到了非模式物种, 此外, BSA与RNA结合的BSR-seq技术也能应用于一些未获得参考基因组的物种进行基因定位。BSA-seq技术在挖掘重要农艺性状基因的研究中越来越广泛, 在许多作物中得到广泛的应用, 如水稻定位耐冷候选基因[12]、大豆定位株高和主茎节数候选基因[13]和黄瓜定位抗静脉黄化病毒候选基因[14]。目前, BSA-seq技术在豌豆中的报道较少, 2018年, Zheng等[15]利用SLAF测序, QTL定位与BSA相结合, 在未获得参考基因组的情况下, 利用水稻的参考基因组与获得的reads进行比对, 检测到2个与叶形性状相关的QTL。
豌豆的基因组较大(约为4.45 Gb), 国内外研究团队较少, 导致豌豆基因组研究有所滞后。2019年, 豌豆首个参考基因组的发布[16], 使得克隆重要农艺性状基因进行基因功能研究变得具有可操作性。本研究以豌豆G0002930、G0004562及其杂交后代, 利用BSA-seq技术与豌豆参考基因组对比并设计InDel分子标记, 验证BSA-seq技术在快速高效定位豌豆花色基因中的可行性, 期望能为豌豆其他基因的精细定位提供思路与参考, 加速豌豆其他重要农艺性状如抗病、抗虫等基因的挖掘, 促进豌豆育种发展。
1 材料与方法
1.1 材料来源
试验材料由国家作物种质库提供: 母本G0002930, 白花, 来源于美国; 父本G0004562, 紫花, 来源于中国西藏。
1.2 研究群体的构建
1.2.1 杂交后代群体的构建与基因组DNA的提取
以G0002930为母本、G0004562为父本杂交, 获得F1代籽粒, 2019年春季在河北省张家口市沽源县播种F1代, 夏季收获F2种子, 2019—2020年冬季在北京昌平温室种植F2代, 并在开花期观察记录各代群体花色的表型。利用卡方检验对豌豆花色性状的遗传分析进行适合性检验, 公式为
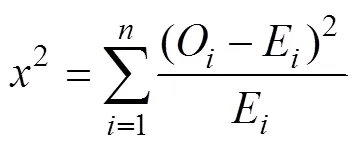
式中,为实际值,为理论值。取植株顶端幼嫩叶片, 与硅胶混合后带回实验室, 采用CTAB法[17]提取豌豆的基因组DNA并检测浓度。
1.2.2 混池的构建 亲本混池构建: 10株白花亲本G0002930与10株紫花亲本G0004562, 命名为R125、R127; 子代混池构建: 表型为白色花和紫色花的F2群体各30株, 命名为R135、R138。DNA等量混合构成2个亲本混池和2个子代混池送与北京百迈客生物科技有限公司完成测序分析, 利用DNA超声波粉碎成插入长度为350 bp的片段, 双端处理文库质检合格后利用Illunima HiSeq进行测序, 亲本深度为10´, 子代测序深度为30´。
1.3 测序数据处理
原始测序序列Raw Reads需筛选过滤, 利用bwa[18]软件将获取高质量的Clean Reads与参考基因组version 1a上[15]比对定位, 利用Picard[19]软件检测插入片段的大小分布, GATK[20]软件实现SNP的变异检测, SnpEff[21]软件注释Small InDel位点。检测到的SNP以及InDel位点过滤掉低质量可信位点。最后利用欧氏距离(euclidean distance, ED)算法[22]、SNP-index (InDel-index)算法[23], 进行关联分析。
1.4 豌豆花色基因的精细定位
利用候选区域的InDel位点设计引物, 在Pisum-URGI网站(https://urgi.versailles.inra.fr/)截取豌豆参考基因组序列变异位点上下游400 bp的序列,利用NCBI的Primer-BLAST工具设计引物, 由生工生物工程(上海)股份有限公司合成引物。亲本PCR反应体系是20 μL, 群体PCR反应体系是10 μL。反应程序如下: 预变性95℃ 5 min; 变性94℃ 30 s, 退火50~60℃ 30 s, 延伸72℃ 45 s, 35个循环; 延伸72℃ 10 min。利用8%的PAGE凝胶电泳对扩增产物进行检测, 统计条带, 计算交换率。
1.5 候选区域功能注释与候选基因筛选
利用SNP和InDel关联分析整合得到的候选区域, 运用BLAST软件与NR数据库[24]、Swissprot数据库、GO数据库[25]、KEGG[25]和COG[27]数据库进行比对, 通过对基因编码区的注释筛选出候选基因。
2 结果与分析
2.1 花色性状的遗传分析
用表型为白花的G0002930作为母本, 与表型为紫花的G0004562杂交, 获得F1代, F1表现为紫花, 表明紫花性状为显性, 将收获的F2种子种于北京昌平温室, 共收获F2群体1262株, 其中紫花群体941株, 白花群体321株。经卡方检验发现(表1), χ2= 0.13 < χ2(0.05,1)= 3.84, 故应接受该花色性状分离符合孟德尔3∶1的分离定律。通过遗传性分析可知, 白花性状受1对隐性核基因控制。
2.2 花色基因初定位区间的筛选
原始测序序列Raw Reads经筛选过滤, 获得Clean Data共435.06 Gbp, 质量值大于30的碱基数占80%以上, 平均每个样品的测序深度为27.89×。基因组平均覆盖度为84.60%, 样品与参考基因组平均比对效率在99%以上(表2), 样品的平均比对效率均在80%以上, 说明样品测序正常, 可用于后续相关研究。
利用测序结果通过关联分析筛选花色基因的候选区间。其中, 基于SNP位点分析(图1), 利用ED算法根据关联阈值判定, 共获得24个与候选基因相关的定位区间(长度总计142.78 Mb), 区间内共有1652个基因, 非同义突变的基因占24.76%; 为了充分利用数据, 降低SNP-index算法中的关联阈值以期获得比较可能的定位区域, 本研究将关联阈值调至0.42, 获得了10个与候选基因相关联的区域(长度总计为32.25 Mb), 区间内包含462个基因, 非同义突变的基因占26.19%。

表1 F2群体中不同花色的植株数目

表2 不同样品测序比对结果

图1 关联值在染色体上的分布(SNP)
A: ED关联分析结果。横坐标为染色体的分布, 每个点代表SNP位点的ED值。B: SNP-index关联分析结果。横坐标为染色体的分布, 每个点代表的ΔSNP-index值。
A: ED correlation analysis. The horizontal coordinate is the distribution of chromosomes, and each point represents the ED value of SNP. B: SNP-index association analysis. The horizontal coordinate is the distribution of chromosomes, and each dot represents the ΔSNP-index value.
基于InDel位点分析(图2), ED算法共得到53个区域(总长度为140.20 Mb), 区域内共包含1523个基因, 包括64个移码突变的基因; InDel-index算法共得到10个区域(总长度为32.27 Mb), 区域内共包含458个基因, 包括16个移码突变的基因。
综上, 取SNP和InDel位点关联分析结果的重复区域作为花色基因初定位的候选区域, 共得到的5个关联区域, 总长度为31.42 Mb, 共包含基因449个(表3)。
2.3 花色基因的精细定位
根据BSA-seq的测序数据, 在初定位31.42 Mb的区间内共设计了279对引物, 利用亲本混合样、2个亲本以及随机选取若干F2群体筛选出条带清晰且有多态性的高质量引物31对, 多态性引物见表4。
利用F2子代群体内321株表型为白花的全部隐性单株和31对多态性的引物进行连锁标记筛选。如图3所示, 在候选区域内共检测到2种交换株, 其中标记WDBH141-1和WDBH187-1共检测到14株交换株和2株交换株, 标记WDBH044-1和WDBH141-1共检测到17株交换株和1株交换株, 标记WDBH209-1和WDBH029-1检测到的交换单株株数为0, 其他标记检测到的交换株详见附表1。故精细定位将最初的31.42 Mb逐步缩小至标记WDBH187-1和WDBH220-1之间, 标记的位置为6,757,954,183和68,566,772, 区间大小0.99 Mb, 共包含19个基因。
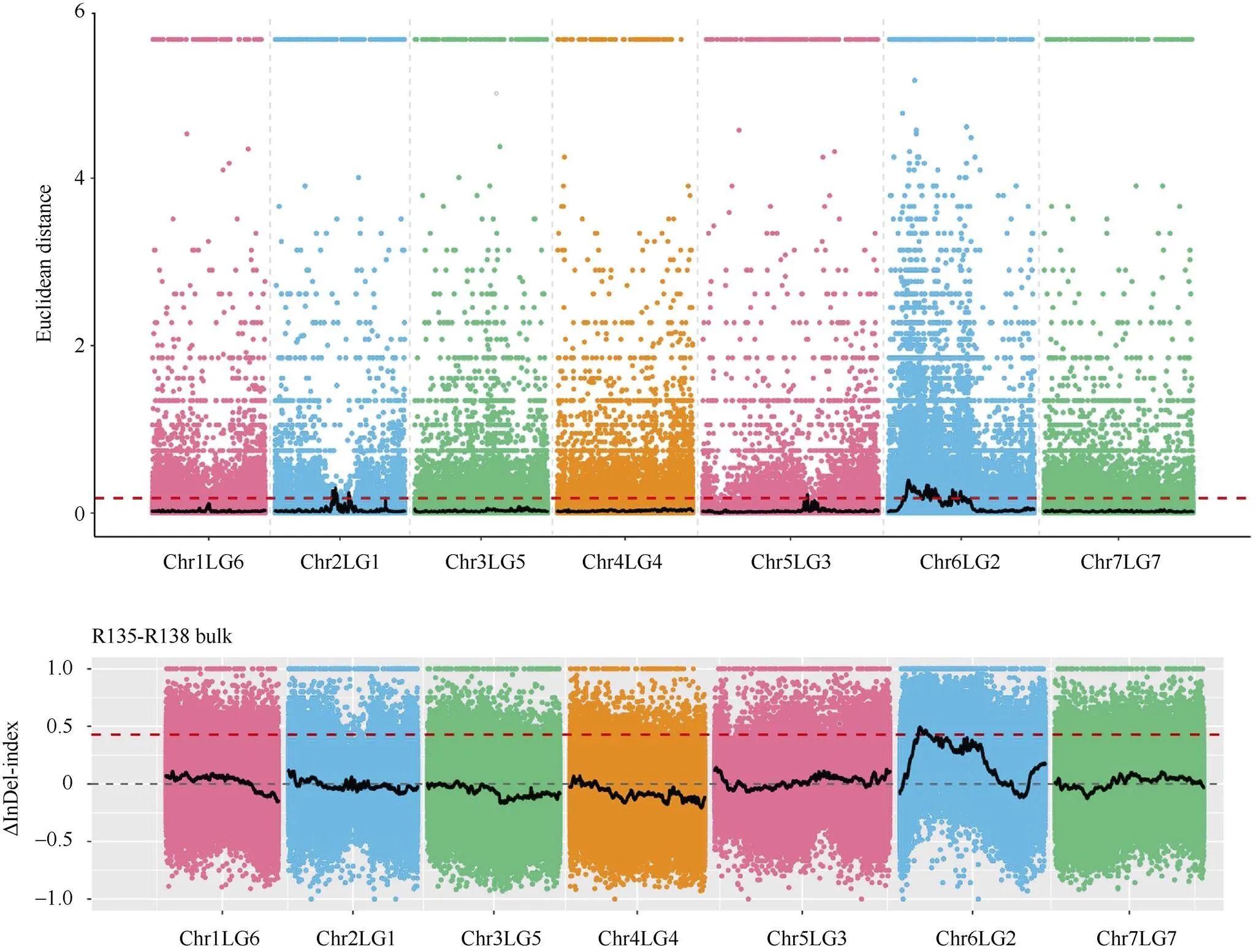
图2 关联值在染色体上的分布(InDel)
A: ED关联分析结果。横坐标为染色体的分布, 每个点代表InDel位点的ED值。B: SNP-index关联分析结果。横坐标为染色体的分布, 每个点代表的ΔInDel-index值。
A: ED correlation analysis. The horizontal coordinate is the distribution of chromosomes, and each point represents the ED value of InDel site. B: SNP-index association analysis. The horizontal coordinate is the distribution of chromosomes, and each dot represents the ΔInDel-index value.

表3 不同关联分析方法获得的关联区域
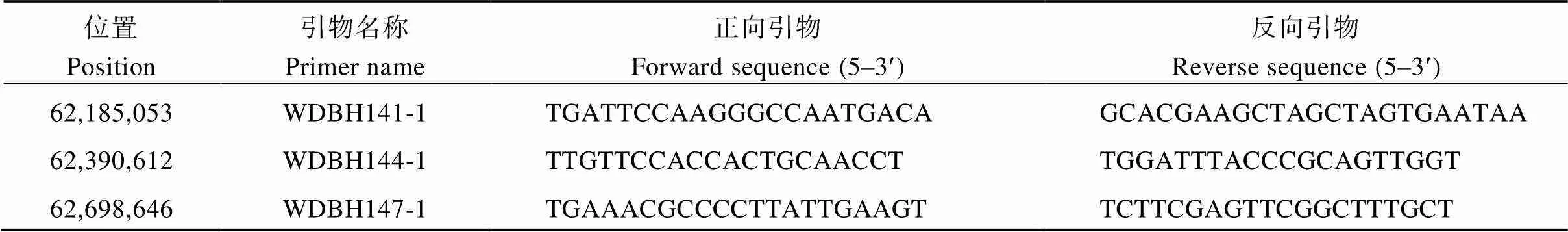
表4 精细定位所用多态性分子标记
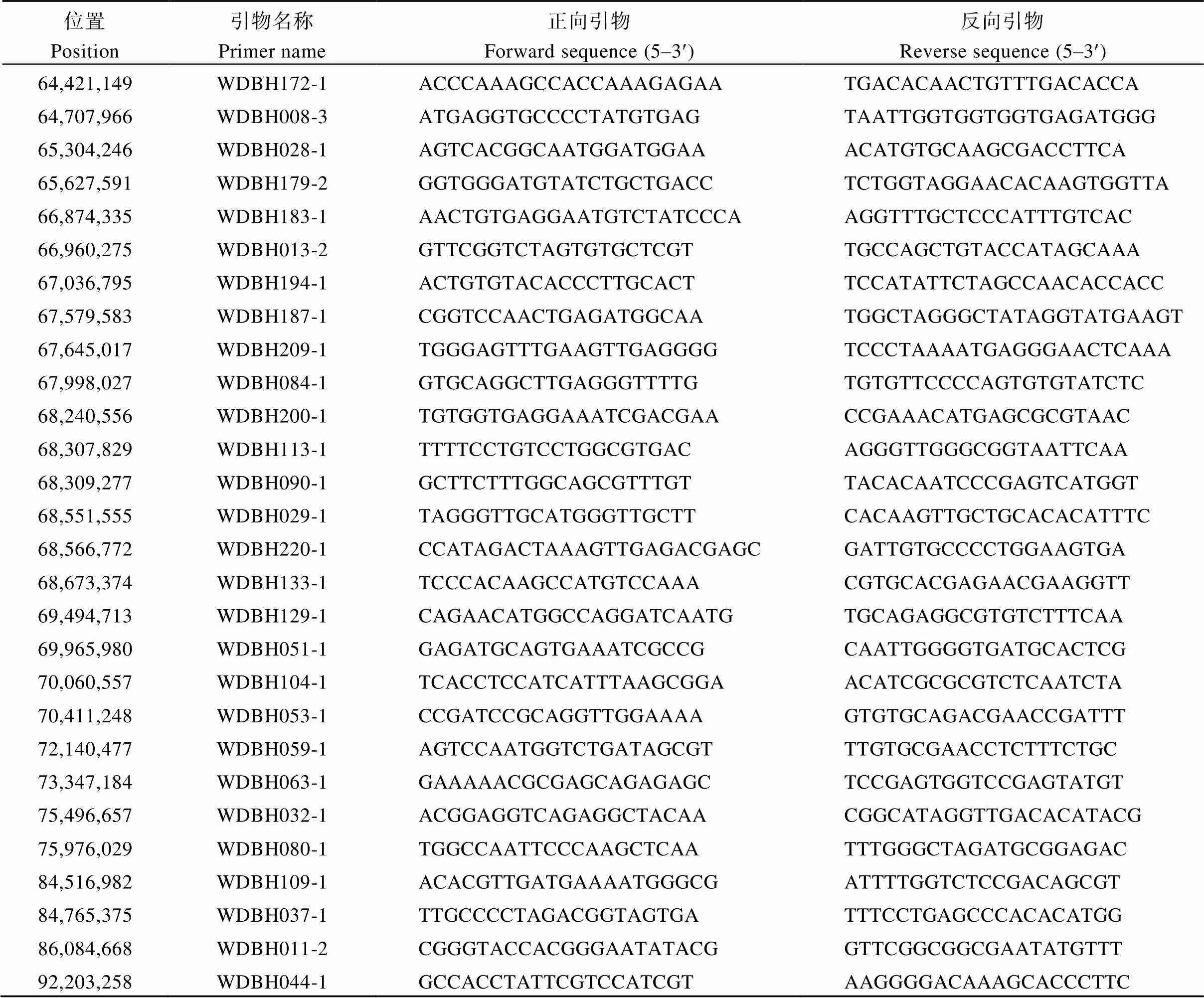
(续表4)

图3 精细定位豌豆花色基因的目标区域
2.4 候选基因及功能预测
由表5可知, 在0.99 Mb的区间内共包含候选基因19个, 其中基因、、在Swissprot_annotation基因注释文库里的功能未知, 基因、、、在所有的基因注释文库里未找到相应的注释, 推测可能是豌豆特有的新基因或者是假基因。其中基因控制Basic helix-loop-helix protein A的合成, 为bHLH转录调控因子, 调控黄酮生物合成过程, 推测可能为豌豆花色的候选基因。
3 讨论
与同源克隆和基于构建遗传图谱QTL定位相比, BSA-seq技术只需少量极端个体构建混池, 混池间除了目标性状相关位点存在差异, 其他位点均趋向于一致, 具有简单、高效、快速定位基因且无需构建遗传图谱等优势[28]。检测结果还提供了区间内突变位点的信息, 在精细定位过程中, 能为分子标记的设计提供了参考和便利。Zheng等[15]通过BSA-seq技术对豌豆进行混池测序, 共获得了67.85 Mb reads,利用水稻参考基因组序列比对得到了0.97 Mb的reads, 与本研究获得的数据相比较数据量较少。本研究利用BSA-seq技术对控制豌豆花色基因进行初步定位, 获得定位区间大小为31.42 Mb, 区间内包含449个基因, 区间长度较大, 可能的原因推测为豌豆的基因组较大(4.45 Gb), 是豆科模式植物蒺藜苜蓿[29](0.5 Gb)的9倍, 但是基因的数量却略少于蒺藜苜蓿, 这也解释了31.42 Mb的范围内仅含有449个基因的原因。在利用ED算法进行关联分析时, 关联阈值计算的0.42, 并未超过理论关联阈值0.667, 可能的原因是构建群体所用的亲本为豌豆种质资源,背景复杂, 噪点较多, 但并不影响候选基因定位的准确性。这种现象在东乡野生稻种子耐贮性相关位点的鉴定[30]和萝卜基因的突变使花瓣颜色从紫色变为白色[31]等相关研究上均有出现。
豌豆的花瓣呈现紫色是由于花瓣中花青素的积累导致的, 花青素是类黄酮途径特定分支的最终产物, 其合成途径在玉米[32]、矮牵牛[33]、葡萄[34]等高等植物中是保守的。在拟南芥中, 花青素的合成需要MYB-bHLH-WD40转录因子的三元复合物(MBW)的调节, 即R2R3-MYB结构域、bHLH (Basic helix- loop-helix protein, 碱性螺旋-环-螺旋)结构域和保守的WD40重复序列的蛋白质家族成员[35]。bHLH转录因子在多种植物中被证实参与了调控花青素合成基因的表达[36], 通过直接调节结构基因(、、)的表达水平, 在花青素途径中起正或负调节作用。在蒺藜苜蓿中, bHLH转录因子被表征为控制花青素和原花青素生物合成的三元复合物的核心成分,突变体导致花青素和原花青素损失,基因对拟南芥突变体的互补能恢复花青素和原花青素的积累[37], 在豌豆中, Hellans等[38]利用模型豆科植物蒺藜苜蓿的参考基因组序列以及它们与豌豆基因组的已知共线性、等位基因多样性、转录量化和瞬时表达互补研究来鉴定豌豆中的基因, 证实了基因不直接参与花青素的合成, 而是编码一个转录因子bHLH参与调控类黄酮化合物合成,基因突变后导致mRNA的剪切错误导致翻译提前终止。本研究利用BSA-seq方法以及设计InDel分子标记进行精细定位, 最后将花色基因定位在0.99 Mb的区间内, 包含19个基因, 其中Swissprot注释库里基因控制Basic helix-loop- helix protein A的合成, 为bHLH转录调控因子。在GO注释库里, 该基因的生物过程包括含花青素的化合物生物合成过程(GO: 0009718)和黄酮生物合成过程的调控(GO: 0009962), 这与Hellans等[38]的研究结果一致, 使得将BSA-seq技术应用到豌豆控制花色基因的定位上得到验证, 为豌豆有关产量、抗虫、抗病等其他重要基因的挖掘奠定了基础, 也为后续基因的功能验证奠定基础。

表5 定位区间内的基因及功能预测
4 结论
本研究利用BSA-seq技术、设计InDel分子标记将花色基因定位到0.99 Mb的区间内, 其中包含19个基因, 通过基因注释文库信息推测为花色候选基因, 为快速高效定位豌豆其他重要性状相关基因奠定了基础。
[1] Pandey A K, Rubiales D, Wang Y, Fang P, Sun T, Liu N, Xu P. Omics resources and omics-enabled approaches for achieving high productivity and improved quality in pea (L.)., 2021, 134: 755–776.
[2] Food and Agriculture Organization of the United Nations. Agriculture production data. https://www.fao.org/faostat/en/#compare.
[3] Fan Z, Zhao Y, Chai Q, Zhao C, Yu A, Coulter J A, Gan Y, Cao W. Synchrony of nitrogen supply and crop demand are driven via high maize density in maize/pea strip intercropping.,2019, 9: 10954.
[4] Humphry M, Reinstädler A, Ivanov S, Bisseling T, Panstruga R. Durable broad-spectrum powdery mildew resistance in peaplants is conferred by natural loss-of-function mutations in., 2011, 12: 866–878.
[5] Hecht V, Laurie R E, Vander Schoor J K, Ridge S, Knowles C L, Liew L C, Sussmilch F C, Murfet I C, MacKnight R C, Weller J L. The peagene is ahomolog necessary for graft-transmissible specification of flowering but not for responsiveness to photoperiod., 2011, 23: 147–161.
[6] Sussmilch F C, Berbel A, Hecht V, Vander Schoor J K, Ferrándiz C, Madueño F, Weller J L. Peais anhomolog that is essential for flowering and compound inflorescence development., 2015, 27: 1046–1060.
[7] Zeng Z B. Precision mapping of quantitative trait loci.,1994, 136: 1457–1468.
[8] Lander E S, Botstein D. Mapping mendelian factors underlying quantitative traits using RFLP linkage maps., 1989, 121: 185–199.
[9] 王建康. 数量性状基因的完备区间作图方法. 作物学报, 2009, 35: 239–245.
Wang J K. Inclusive composite interval mapping of quantitative trait genes., 2009, 35: 239–345 (in Chinese with English abstract).
[10] Wu L, Fredua-Agyeman R, Hwang S F, Chang K F, Conner R L, McLaren D L, Strelkov S E. Mapping QTL associated with partial resistance to Aphanomyces root rot in pea (L.) using a 13.2 K SNP array and SSR markers., 2021, 134: 2965–2990.
[11] Aznar-Fernández T, Barilli E, Cobos M J, Kilian A, Carling J, Rubiales D. Identification of quantitative trait loci (QTL) controlling resistance to pea weevil () in a high-density integrated DArTseq SNP-based genetic map of pea., 2020, 10: 33.
[12] Guo Z, Cai L, Chen Z, Wang R, Zhang L, Guan S, Zhang S, Ma W, Liu C, Pan G. Identification of candidate genes controlling chilling tolerance of rice in the cold region at the booting stage by BSA-seq and RNA-seq., 2020, 7: 201081.
[13] Li R, Jiang H, Zhang Z, Zhao Y, Xie J, Wang Q, Zheng H, Hou L, Xiong X, Xin D, Hu Z, Liu C, Wu X, Chen Q. Combined linkage mapping and BSA to identify QTL and candidate genes for plant height and the number of nodes on the main stem in soybean., 2019, 21: 42.
[14] Pujol M, Alexiou K G, Fontaine A S, Mayor P, Miras M, Jahrmann T, Garcia-Mas J, Aranda M A. Mapping cucumber vein yellowing virus resistance in cucumber (L.) by using BSA-seq analysis., 2019, 10: 1583.
[15] Zheng Y, Xu F, Li Q, Wang G, Liu N, Gong Y, Li L, Chen Z H, Xu S. QTL mapping combined with bulked segregant analysis identify SNP markers linked to leaf shape traits inusing SLAF sequencing., 2018, 9: 615.
[16] Kreplak J, Madoui M A, Cápal P, Novák P, Labadie K, Aubert G, Bayer P E, Gali K K, Syme R A, Main D, Klein A, Bérard A, Vrbová I, Fournier C, d’Agata L, Belser C, Berrabah W, Toegelová H, Milec Z, Vrána J, Lee H, Kougbeadjo A, Térézol M, Huneau C, Turo C J, Mohellibi N, Neumann P, Falque M, Gallardo K, McGee R, Tar’an B, Bendahmane A, Aury J M, Batley J, Le Paslier M C, Ellis N, Warkentin T D, Coyne C J, Salse J, Edwards D, Lichtenzveig J, Macas J, Doležel J, Wincker P, Burstin J. A reference genome for pea provides insight into legume genome evolution., 2019, 51: 1411–1422.
[17] Murray M G, Thompson W F. Rapid isolation of high molecular weight plant DNA., 1980, 8: 4321–4325.
[18] Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform., 2009, 25: 1754–1760.
[19] Source Forge. Picard. San Diego, CA, USA. http://sourceforge. net/projects/picard/.
[20] McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo M A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data., 2010, 20: 1297–1303.
[21] Cingolani P, Platts A, Wang L L, Coon M, Nguyen T, Wang L, Land S J, Lu X, Ruden D M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. SNPs in the genome ofstrain w1118; iso-2; iso-3.(Austin), 2012, 6: 80–92.
[22] Hill J T, Demarest B L, Bisgrove B W, Gorsi B, Su Y C, Yost H J. MMAPPR: mutation mapping analysis pipeline for pooled RNA-seq.,2013, 23: 687–697.
[23] Fekih R, Takagi H, Tamiru M, Abe A, Natsume S, Yaegashi H, Sharma S, Sharma S, Kanzaki H, Matsumura H, Saitoh H, Mitsuoka C, Utsushi H, Uemura A, Kanzaki E, Kosugi S, Yoshida K, Cano L, Kamoun S, Terauchi R. MutMap+: genetic mapping and mutant identification without crossing in rice., 2013, 8: e68529.
[24] Deng Y, Jianqi L I, Songfeng W U, Zhu Y, Chen Y, Fuchu H E. Integrated nr database in protein annotation system and its localization., 2006, 32: 71–72.
[25] Ashburner M, Ball C A, Blake J A, Botstein D, Butler H, Cherry J M, Davis A P, Dolinski K, Dwight S S, Eppig J T, Harris M A, Hill D P, Issel-Tarver L, Kasarskis A, Lewis S, Matese J C, Richardson J E, Ringwald M, Rubin G M, Sherlock G. Gene ontology: tool for the unification of biology. The gene ontology consortium.,2000, 25: 25–29.
[26] Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome.,2004, 32: D277–D280.
[27] Tatusov R L, Galperin M Y, Natale D A, Koonin E V. The COG database: a tool for genome-scale analysis of protein functions and evolution., 2000, 28: 33–36.
[28] Gillmor C S, Roeder A H, Sieber P, Somerville C, Lukowitz W. A genetic screen for mutations affecting cell division in theembryo identifies seven loci required for cytokinesis., 2016, 11: e0146492.
[29] Branca A, Paape T D, Zhou P, Briskine R, Farmer A D, Mudge J, Bharti A K, Woodward J E, May G D, Gentzbittel L, Ben C, Denny R, Sadowsky M J, Ronfort J, Bataillon T, Young N D, Tiffin P. Whole-genome nucleotide diversity, recombination, and linkage disequilibrium in the model legume., 2011, 108: E864–E870.
[30] Zhao M, Hu B, Fan Y, Ding G, Yang W, Chen Y, Chen Y, Xie J, Zhang F. Identification, analysis, and confirmation of seed storability-related loci in dongxiang wild rice (Griff.).(Basel), 2021, 12: 1831.
[31] Liu D, Wei X, Sun D, Yang S, Su H, Wang Z, Zhao Y, Li L, Liang J, Yang L, Zhang X, Yuan Y. An SNP mutation of gene RsPP converts petal color from purple to white in radish (L.)., 2021, 12: 643579.
[32] Qin L, Sun L, Wei L, Yuan J, Kong F, Zhang Y, Miao X, Xia G, Liu S. Maize SRO1e represses anthocyanin synthesis through regulating the MBW complex in response to abiotic stress., 2021, 105: 1010–1025.
[33] Albert N W, Lewis D H, Zhang H, Schwinn K E, Jameson P E, Davies K M. Members of an R2R3-MYB transcription factor family inare developmentally and environmentally regulated to control complex floral and vegetative pigmentation patterning., 2011, 65: 771–784.
[34] He F, Mu L, Yan G L, Liang N N, Pan Q H, Wang J, Reeves M J, Duan C Q. Biosynthesis of anthocyanins and their regulation in colored grapes.,2010, 15: 9057–9091.
[35] Petroni K, Tonelli C. Recent advances on the regulation of anthocyanin synthesis in reproductive organs.,2011, 181: 219–229.
[36] Deng J, Li J, Su M, Lin Z, Chen L, Yang P. A bHLH geneofregulates anthocyanin biosynthesis., 2021, 158: 518–523.
[37] Li P, Chen B, Zhang G, Chen L, Dong Q, Wen J, Mysore K S, Zhao J. Regulation of anthocyanin and proanthocyanidin biosynthesis bybHLH transcription factor., 2016, 210: 905–921.
[38] Hellens R P, Moreau C, Lin-Wang K, Schwinn K E, Thomson S J, Fiers M W, Frew T J, Murray S R, Hofer J M, Jacobs J M, Davies K M, Allan A C, Bendahmane A, Coyne C J, Timmerman- Vaughan G M, Ellis T H. Identification of Mendel’s white flower character., 2010, 5: e13230.

Fine mapping of flower colour gene in pea (L.) based on BSA-seq technique
YAN Xin1,**, XIANG Chao2,**, LIU Rong1, LI Guan1, LI Meng-Wei1, LI Zheng-Li3, ZONG Xu-Xiao1,*, and YANG Tao1,*
1Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, Beijing 100081, China;2Institute of Crop Sciences, Sichuan Academy of Agricultural Sciences, Chengdu 610066, Sichuan, China;3Institute of Horticulture, Guizhou Academy of Agricultural Sciences, Guiyang 550006, Guizhou, China
In recent years, BSA-seq technology has been widely used in the mining of new genes related to agronomic traits. With the development of the first reference genome of pea, it is imperative to combine BSA-seq method with genome-wide sequencing for gene mapping. In this study, we used purple flower parent G0004562, white flower parent G0002930, and F2populations for preliminarily locate the target genes controlling flower color by BSA-seq technology, and a mapping region of 31.42 Mb was obtained. Then, the InDel molecular markers were designed to further narrow the mapping interval, and finally the target gene was located in the range of 0.99 Mb with 19 genes. Based on gene annotation,was considered as the candidate gene that controled the flower color. The results of this study verified the feasibility of gene mapping by BSA-seq technology in pea.
pea; BSA-seq; InDel; gene mapping
10.3724/SP.J.1006.2023.24055
本研究由国家作物种质资源库-食用豆资源整合与共享项目(NCGRC-2021-07), 普查收集食用豆资源鉴定评价与繁殖编目入库项目(19210867)和财政部和农业农村部国家现代农业产业技术体系建设专项(CARS-08-G11)资助。
This study was supported by the National Infrastructure for Crop Germplasm Resources Project from the Ministry of Science and Technology of China (NCGRC-2021-07), the Project of Identification, Evaluation, Replication, and Preservation of Food Legumes Collected by the Survey and Collection Action on Crop Germplasm Resources (19210867), and the China Agriculture Research System of MOF and MARA (CARS-08-G11).
杨涛, E-mail: yangtao02@caas.cn; 宗绪晓, E-mail: zongxuxiao@caas.cn
**同等贡献(Contributed equally to this work)
严昕, E-mail: yanxin5290@163.com; 项超, E-mail: xc2011cib@163.com
2022-03-10;
2022-07-21;
2022-08-22.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20220819.1436.008.html
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
