基于实验室能力验证需求国标法检测鸡肉和鸡蛋中氟喹诺酮类药物残留量的优化
2023-02-13唐子恒林泽宁陈子敏黄楚彪劳华杰詹炜玮刘文字沈祥广
唐子恒,林泽宁,陈子敏,黄楚彪,劳华杰,詹炜玮,刘文字,沈祥广*
(1.华南农业大学国家兽药残留基准实验室,广东广州 510642)(2.农业农村部畜禽产品质量监督检验测试中心(广州),广东广州 510642)
畜禽产品中兽药及禁用药物残留检测能力验证(以下简称“能力验证”)是指各个实验室或检测机构按照事先制定好的标准与规定,对由负责单位事先准备好的考核样品进行检测,通过对比各实验室或检测机构的结果与操作规范性,来评价该实验室或检测机构的兽药及禁用药物残留检测能力[1,2]。根据《检验检测机构资质认定能力评价检验检测机构通用要求》和《农产品质量安全检测机构考核评审细则》的要求[3,4],检验检测机构及相关实验室必须参加能力验证或实验室间比对。由于未通过能力验证可能导致实验室或检测机构失去相应项目的检测活动及出具合法有效检验检测报告的资质,所以尽可能地提高能力验证通过率对于实验室或检测机构来说十分重要。
氟喹诺酮类药物(Fluoroquinolones,FQs)是广谱合成抗生素[5],可以选择性抑制参与革兰氏阴性菌和革兰氏阳性菌DNA复制的细菌酶[6]。因为它们对多种细菌病原体都具有活性,且与其他抗菌药无交叉耐药性[7],加之价格低廉,故常被用于大规模养殖,以预防、治疗动物疾病或改善畜禽生长和生产力[8,9]。由此,不规范用药带来的残留风险也较大,它们会残留在动物体内,并通过食物链进入人体,对人体健康造成极大危害[10]。FQs在畜牧兽医领域应用的不良反应鲜有报道,但对于人类来说,FQs存在中枢系统毒性,会导致如Q-T间期延长,肌肉疼痛无力,肌腱撕裂等不良反应[11]。从20世纪90年代中期开始,革兰氏阳性菌株和革兰氏阴性菌株对 FQs的耐药性不断增加[12,13]。近年来,人们还发现重要的水资源中也存在FQs残留[14],药物残留已经威胁到人类饮用水安全。
根据GB 31650-2019《食品安全国家标准食品中兽药最大残留限量》规定[15],鸡肉中的达氟沙星(Danofloxacin,DAN)最大残留限量(Maximum Residue Limit,MRL)为 200 µg/kg,沙拉沙星(Sarafloxacin,SAR)MRL 为 10 µg/kg,恩诺沙星(Enrofloxacin,ENR)与环丙沙星(Ciprofloxacin,CIP)两者残留量相加不超过100 µg/kg。农业农村部近年来组织的能力验证常将鸡肉中该4种药物残留量测定作为考核项目。上述4种FQs药物在蛋鸡的产蛋期禁用。
进行能力验证考核时,存在样品数量少、检测时间限制等难点。针对这些难点,在农业部1025号公告-14-2008和农业部781号公告-6-2006两个现行有效的国标高效液相色谱(HPLC)检测方法的基础上[16,17],增加了移取1 mL待SPE净化的备用液(1 mL)进行加热沉淀净化,并上机检测的快速分析方法,为依照国标方法检测的能力验证实验结果提供质量验证参比。剩余备用液分取适量体积,继续按国标方法优化后的净化操作完成能力验证实验。另外,由于高效液相色谱仪相对价廉,普及率高,所建的简约前处理快速检测法也适合养殖企业产品上市前的自检。
1 材料与方法
1.1 仪器与材料
Agilent 1260高效液相色谱仪(配荧光检测器),美国 Agilent公司;AT-261型电子分析天平,瑞士Mettler公司;DK-8D三孔电热恒温水槽,上海齐欣科学仪器有限公司;KS 501 digital振荡摇床,德国IKA公司;ST 16R高速冷冻离心机,美国赛默飞世尔科技有限公司。
环丙沙星、达氟沙星、恩诺沙星、沙拉沙星四种对照品,中国兽医药品监察所;磷酸二氢钾(分析纯),国药集团化学试剂有限公司;正己烷(分析纯),国药集团化学试剂有限公司;三乙胺(分析纯),上海麦克林生化科技有限公司;磷酸(分析纯),天津市富宇精细化工有限公司;氢氧化钠(分析纯),广州化学试剂厂;乙腈(色谱纯),赛默飞世尔科技有限公司。TC-C18液相色谱柱(250 mm×4.6 mm),安捷伦科技有限公司;C18固相萃取柱(100 mg~1 mL),安捷伦科技有限公司。
空白鸡肉、空白鸡蛋均为市售合格产品,无FQs残留,匀浆后封存于-20 ℃备用。
1.2 实验方法
1.2.1 标准溶液和试剂的配制
标准溶液和试剂参照农业部1025号公告-14-2008和农业部 781号公告-6-2006两个国标检测方法配制[16,17]。
标准储备溶液:精密称取达氟沙星标准品10 mg(按有效成分折算),用0.03 mol/L氢氧化钠溶液溶解,并定容至50 mL得0.2 mg/mL的标准储备液;精密称取恩诺沙星、环丙沙星和沙拉沙星标准品各50 mg(均按有效成分折算),用0.03 mol/L氢氧化钠溶液溶解,并定容至50 mL得1 mg/mL的标准储备液。标准储备溶液置4 ℃冰箱中保存。
混合标准工作液:准确量取适量上述4种储备液混合,用流动相稀释定容成适宜浓度的混合标准工作液,使达氟沙星、恩诺沙星、环丙沙星和沙拉沙星浓度比为1:5:5:5,于4 ℃冰箱保存。如无特殊说明,文中所述的混合标准溶液、添加浓度组中的达氟沙星浓度均为其他药物浓度的1/5。
磷酸盐提取液:取磷酸二氢钾6.8 g,加水溶解并稀释至500 mL,用5.0 mol/L氢氧化钠溶液调节pH值至7.0。
0.05 mol/L磷酸/三乙胺溶液:取浓磷酸3.4 mL,用水稀释至1 000 mL,用三乙胺调pH值至2.4。
乙腈(30%)-缓冲液:量取乙腈30 mL,0.05 mol/L磷酸/三乙胺溶液70 mL,混合,现配现用。
1.2.2 色谱条件
色谱柱:Agilent TC-C18,250 mm×4.6 mm,5 μm;流动相:0.05 mol/L磷酸/三乙胺溶液-乙腈(81:19);流速:0.8 mL/min(鸡肉样品检测),1.0 mL/min(鸡蛋样品检测);进样量:50 μL;检测波长:激发波长280 nm,发射波长450 nm。
1.2.3 标准曲线的制备
准确量取适量混合标准工作液,用流动相稀释成浓度分别为0.5、1.0、5.0、10.0、20.0、200.0、400.0 µg/L的混合标准溶液(其中达氟沙星浓度为0.1、0.2、1.0、2.0、4.0、40.0、80.0 µg/L),供高效液相色谱分析,以与样品同色谱条件重复检测三次后取峰面积平均值。以各药物浓度(µg/L)为横坐标(x轴),对应峰面积平均值为纵坐标(y轴),进行线性回归分析,计算回归方程和相关回归系数。
1.2.4 样品提取
鸡肉:称取(2±0.05)g试样于50 mL离心管中(如为考核样品,默认样品质量为2.00 g),在样品中精确加入磷酸盐提取液10.0 mL,涡旋1 min后,将离心管盖紧,置摇床振荡10 min,振荡结束放入高速离心机10 000 r/min离心5 min。离心后的上清液转移至另一50 mL离心管,残余物再用10 mL磷酸盐提取液同法提取。合并上清液并混匀,样液备用。
鸡蛋:称取(2±0.05)g试样于50 mL离心管中(如为考核样品,默认样品质量为2.00 g),精确加入磷酸盐提取液2.0 mL,涡旋1 min后,将离心管盖紧,置摇床振荡 10 min,振荡结束放入高速离心机10 000 r/min离心5 min。离心后的上清液转移至10 mL量筒,残余物再用2 mL磷酸盐提取液同法提取。合并上清液后,用纯水将上清液定容至6 mL,混匀,样液备用。
1.2.5 样液的净化处理
1.2.5.1 加热快速沉淀法
精密移取备用样液1 mL于2 mL塑料离心管中,准确加入乙腈0.2 mL,盖紧,用封口膜密封,涡旋30 s,置55 ℃水浴加热20 min后,4 ℃条件下以15 000 r/min离心5 min,上清液过0.22 µm滤膜后进高效液相色谱仪快速测定样品中的药物含量。
1.2.5.2 按农业部1025号公告-14-2008方法净化的优化方法
鸡肉样品备用液先放入-20 ℃冰箱冷冻至结冰,重解冻后10 000 r/min离心5 min,精密移取上层清夜5 mL,加入经甲醇、磷酸盐缓冲液各2 mL平衡过的C18固相萃取柱中,过柱后,1 mL水淋洗,挤干。用流动相1 mL洗脱,挤干,收集洗脱液,过0.22 µm滤膜后进高效液相色谱仪测定。
1.2.5.3 按农业部781号公告-6-2006方法净化的优化方法
鸡蛋样品备用样液转移至50 mL离心管中,加入水饱和的正己烷10 mL,置水平摇床,振荡10 min,4 ℃条件下以10 000 r/min离心5 min,用吸管吸去上层的正己烷,保留中间絮凝层;再加入10 mL水饱和正己烷,重复振荡和离心,精密移取下层清液3 mL,加入依次用乙腈、乙腈(30%)-缓冲液和磷酸盐提取液各5 mL平衡过的C18固相萃取柱中,过柱后,5 mL水淋洗,挤干。用1 mL流动相洗脱,挤干,收集洗脱液,过0.22 µm滤膜后进高效液相色谱仪测定。
2 结果与讨论
2.1 方法学评价
2.1.1 标准曲线
对“1.2.3”中各浓度的混合标准溶液分别按“1.2.2”色谱条件的鸡肉、鸡蛋两种不同流速条件进行分析,以标准溶液中药物的浓度为横坐标(x轴),对应色谱峰的峰面积为纵坐标(y轴),绘制标准曲线。四种药物在设定的浓度范围内线性关系良好,相关系数(r2)均能达到0.999 9。具体结果如表1所示。

表1 4种氟喹诺酮类药物的线性范围、回归方程、线性相关系数Table 1 Linear ranges, regression equations, correlation coefficients of four fluoroquinolones
2.1.2 检测限与定量限
取空白的鸡肉、鸡蛋样品添加适量混合标准工作液,制成不同浓度的空白添加样品,按“1.2.4”的提取方法提取,“1.2.5”的加热快速沉淀法净化处理完成后上机分析。以信噪比(S/N)≥3所对应的添加浓度为检测限(LOD),S/N≥10所对应的添加浓度为定量限(LOQ),采用加热快速沉淀法的检测灵敏度结果如表2。

表2 4种氟喹诺酮类药物的检测限、定量限(g/kg)Table 2 LODs and LOQs of four fluoroquinolones
2.1.3 回收率与精密度
鸡肉:共设置3个浓度组,取空白鸡肉,添加混合标准工作液,以CIP为参考,制成浓度分别为20、40、100 µg/kg(相当于农业部 1025号公告-14-2008方法LOD的1倍、2倍、5倍)的空白添加样品,每组设置6个平行样品,2个空白对照,重复3个批次。除样品外,每个浓度组设置1个由流动相配成的标准对照溶液和1个由基质匹配的标准对照溶液,标准对照溶液浓度与对应浓度组的理论上机浓度一致,并以前者为对照计算加标回收率及相对标准偏差(RSD)。使用快速加热沉淀法时,各浓度组具体回收率及RSD如表3所示。
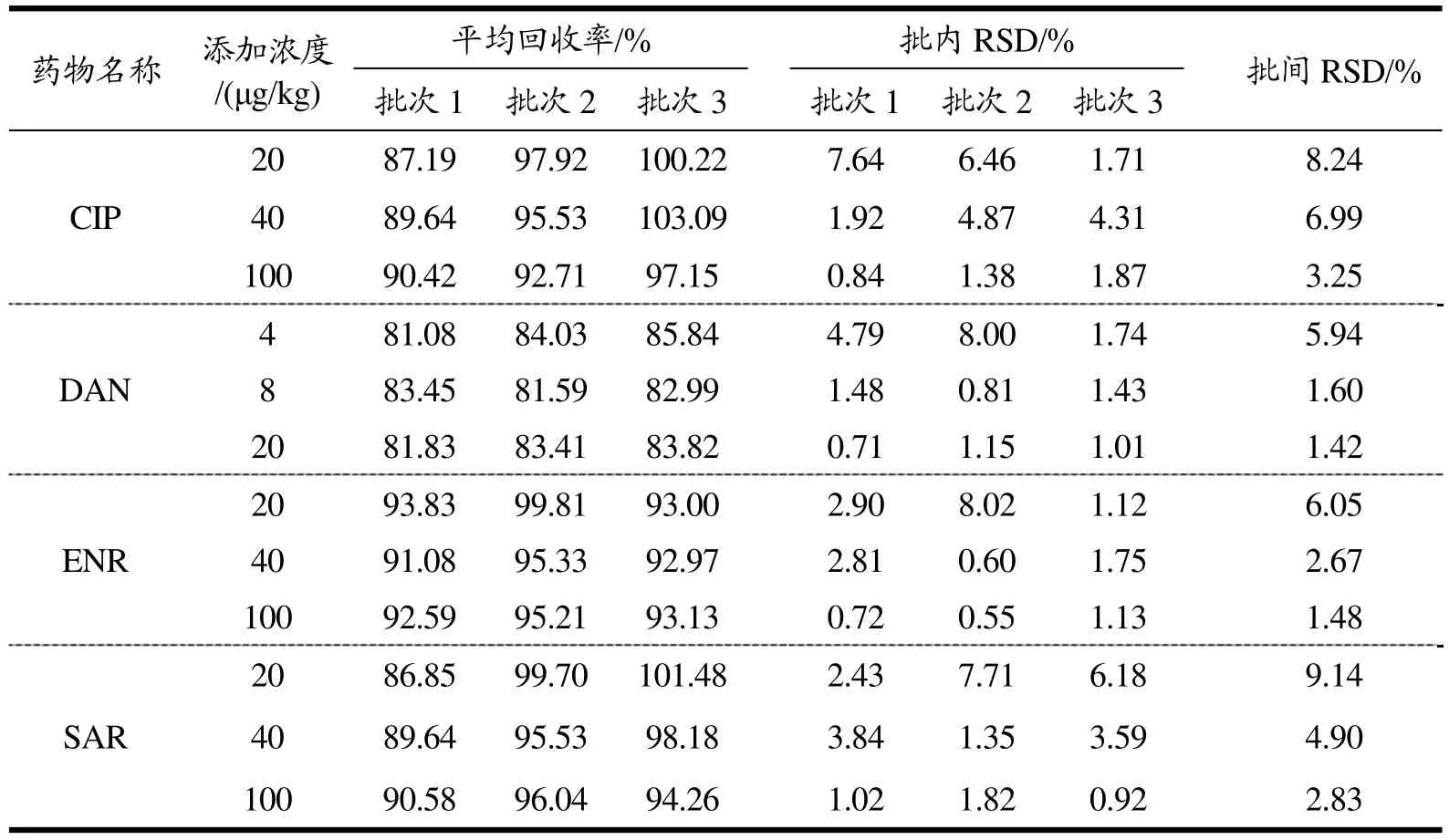
表3 鸡肉中4种氟喹诺酮类药物的回收率及相对标准偏差Table 3 Recoveries, intra-assay and inter-assay RSDs of four fluoroquinolones in chicken
鸡蛋:设置3个浓度组,取空白鸡蛋,添加混合标准工作液,以CIP为参考,制成浓度分别为10 µg/kg、20 µg/kg、50 µg/kg(相当于农业部 781 号公告-6-2006方法LOD的1倍、2倍、5倍)的空白添加样品,每组设置6个平行样品,2个空白对照,重复3个批次。除样品外,每个浓度组设置1个用流动相配成的标准对照溶液和1个由基质匹配的标准对照溶液,标准对照溶液浓度与对应浓度组的理论上机浓度一致,并以前者为对照计算加标回收率及RSD。使用快速加热沉淀法时,各浓度组具体回收率及RSD如表4所示。
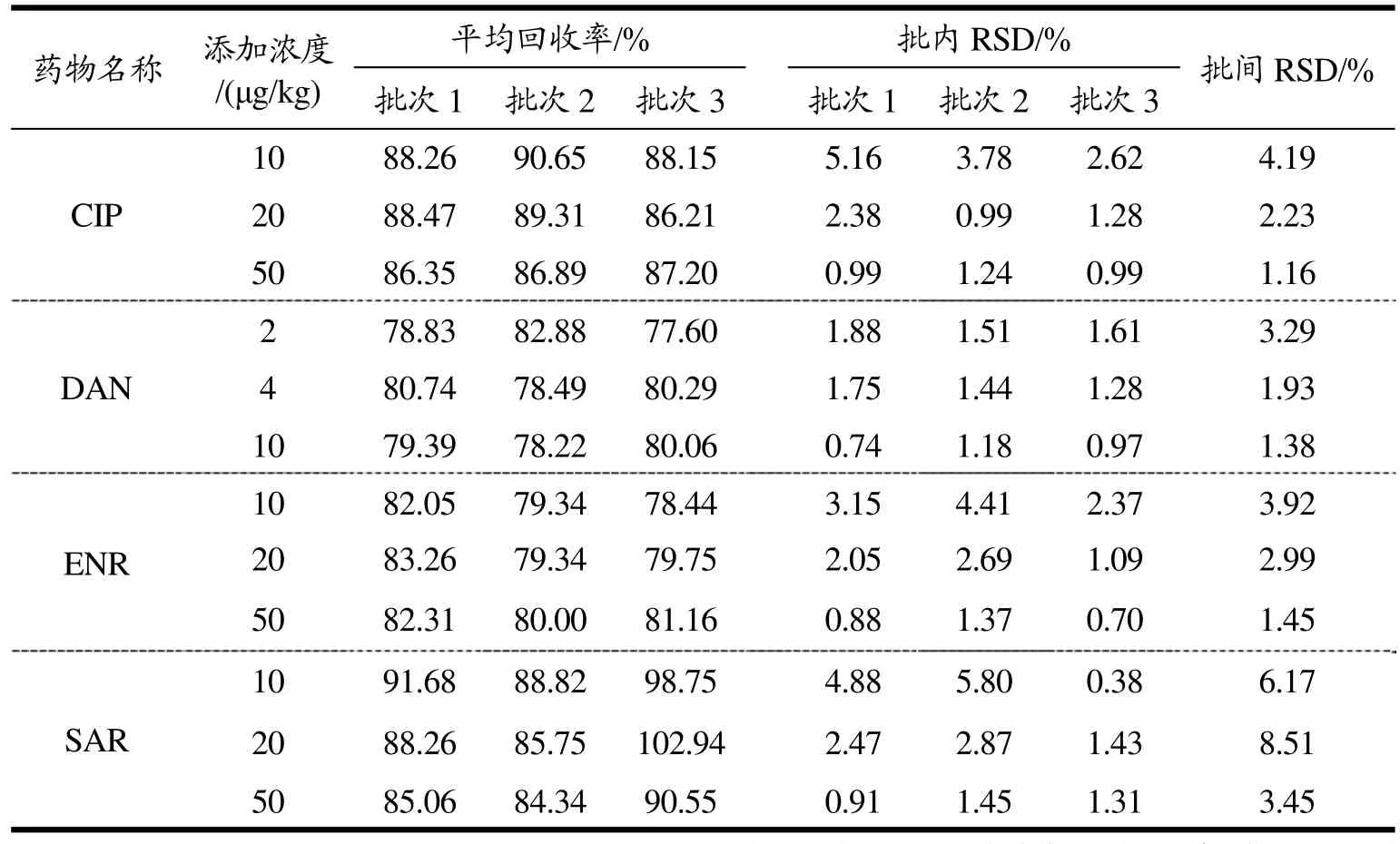
表4 鸡蛋中4种氟喹诺酮类药物的回收率及相对标准偏差Table 4 Recoveries, intra-assay and inter-assay RSDs of four fluoroquinolones in egg
2.2 讨论与分析
2.2.1 色谱条件
农业部781号公告-6-2006和农业部1025号公告-14-2008中使用的流动相均为0.05 mol/L磷酸/三乙胺溶液-乙腈,比例分别为 81:19和 82:18,流速分别为1.0 mL/min和 0.8 mL/min,色谱柱为 C18250 mm×4.6 mm。为使能力验证的工作流程尽量简约,在国标法的流动相条件下,对比了不同型号的色谱柱。GL-sciences Inc Inertsil ODS2 C18、伊利特Hypersil BDS C18和Supelco Discovery C18的药物峰形不够理想;Agilent Extend C18、Waters Nova-Pak C18的药物出峰较早,CIP、DAN和ENR的色谱峰分离不够彻底;相对其他型号色谱柱,Agilent TC-C18色谱柱在等度洗脱条件下对国标中的4种FQs药物有较好的分离效果,药物峰型尖锐、对称,基线平稳,对空白鸡肉和鸡蛋的分析均未见明显干扰峰,每次进样能在20 min以内实现药物的分离和测定。
2.2.2 方法的灵敏度和精密度
快速加热沉淀法虽然是对样品进行了稀释,但由于背景干扰低,通过加大进样量(50 μL),仍能获得较好的检测灵敏度。快速加热沉淀法应用于鸡肉检测时,4种FQs药物的LOD为1 µg/kg~10 µg/kg,LOQ为4 µg/kg~20 µg/kg,添加回收率为81.08%~103.09%;应用于鸡蛋时,4种药物LOD为0.5 µg/kg~5 µg/kg,LOQ为2 µg/kg~10 µg/kg,回收率为77.60%~102.94%,详见表2~表4。灵敏度、准确度、精密度均能满足NY/T 1896-2010《兽药残留实验室质量控制规范》的要求[18],相较于其他文献方法(如表5所示)也具有一定优势。

表5 相关文献方法及其效果Table 5 Methods and effects of relevant literature
鸡肉和鸡蛋定量限添加样品检测色谱图见图1。
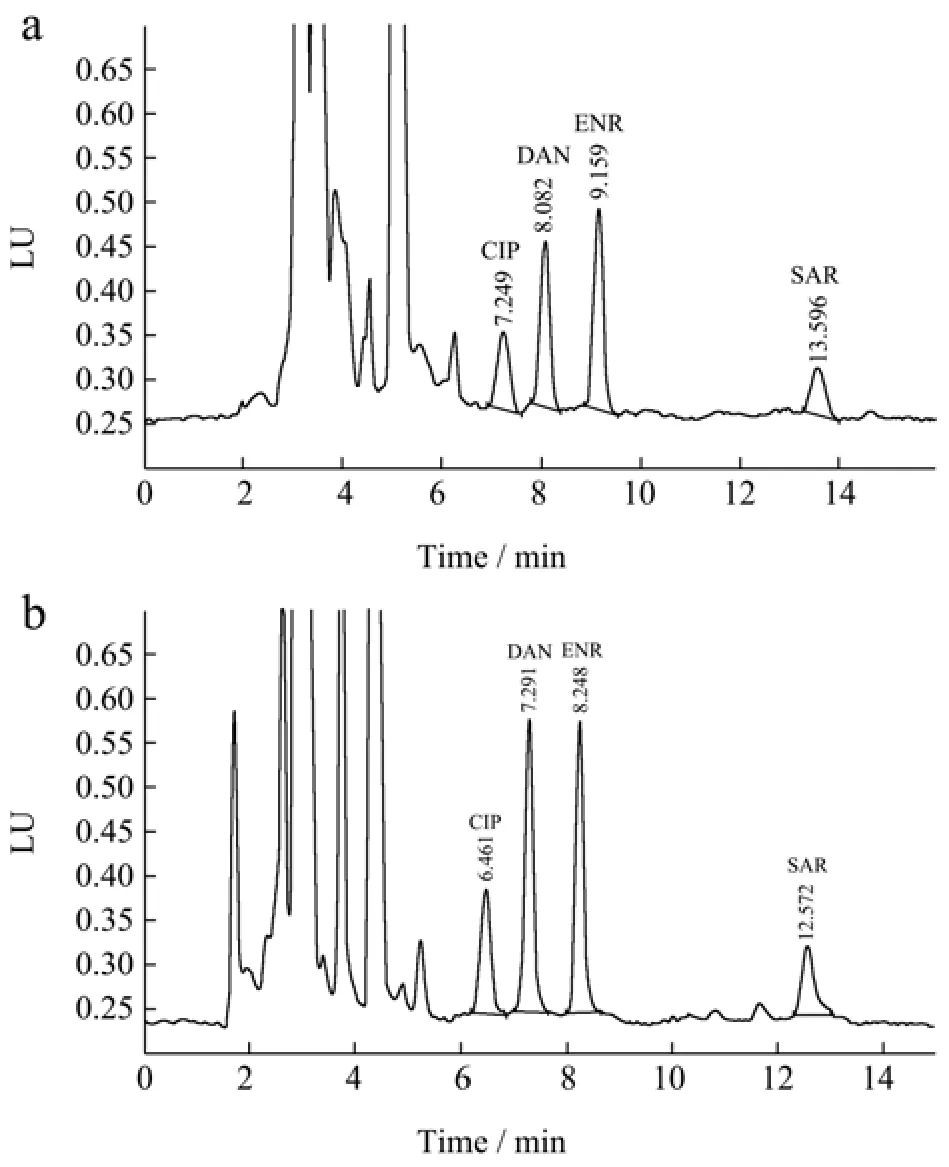
图1 鸡肉及鸡蛋定量限添加样品色谱图Fig.1 HPLC chromatograms of chicken and egg samples with quantitative limit
2.2.3 样品净化方法优化
鸡肉和鸡蛋均含有较高比例的蛋白质,蛋黄中卵磷脂的含量也很高[23],样品的前处理重点在于提取药物并分离蛋白质和脂类等物质,上述两个国标检测方法中均采用pH值7.0的磷酸盐缓冲液作为提取剂,经测试,该方法提取步骤带入的干扰物质少,对氟喹诺酮类药物的提取率较高。但两者的过 SPE柱净化步骤均存在一定缺陷,容易引起堵柱或出现回收率偏低的问题。
加热快速沉淀法主要是利用低含量乙腈结合加热的物理作用使蛋白质等杂质迅速淀沉和分离。温度高于 50 ℃时,蛋白质的凝集效果较好,且温度越高凝集越快,但温度过高则易造成溶剂挥发或药物分解[24]。选用 55 ℃的水浴条件既可以满足沉淀杂质的需求,又不会影响准确定量。若上机样液中的乙腈比例过高,色谱峰会出现前移、峰宽变大或“M”型峰等情况。添加0.2 mL乙腈足够沉淀杂质,样液中乙腈比例与流动相中的乙腈比例相近,过滤后可直接进高效液相色谱仪分析。由于操作简单易控,相对于过SPE柱的净化方式,具有更好的重现性、准确性和过程可控性。加热快速沉淀法只需使用1 mL备用液,不影响样品检测继续按国标方法完成实验和测定。利用加热快速沉淀法对质控样品检测的准确度和精密度评价,并将检测结果与按国标方法完成实验的结果进行比较,能相对可靠的评估按国标方法检测结果的准确性。采用加热快速沉淀法能为参加能力验证实验室多提供一次评估结果准确性的参比机会。
农业部1025号公告-14-2008的净化方法中,虽然只分取5 mL备用液上样进行SPE净化,但在鸡肉样品实际检测中,由于备用液中含有蛋白质和脂肪,堵柱的情况仍常有发生。如果先将过柱备用液置于-20 ℃冻结,使脂肪和蛋白质凝集,重新解冻后高速离心,再取上清液过SPE净化,堵柱和回收率低两个问题皆得到很好的解决,具体回收率对比见图2。浅色部分为按照原方法操作的回收率,其中20 µg/kg浓度组实验中未出现堵柱现象,该份样品为新鲜鸡肉;40 µg/kg和100 µg/kg添加浓度组皆出现了堵柱现象,该份样品为经冻融过的鸡肉。深色部分为按国标的净化方法优化后的回收率,结果较为理想。
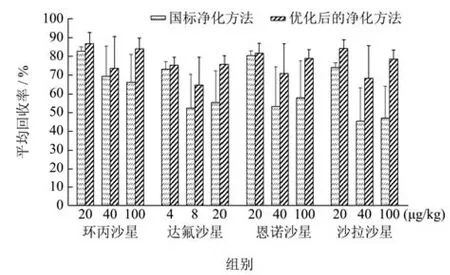
图2 鸡肉样品回收率对比图Fig.2 Comparison of chicken sample recovery
在农业部781号公告-6-2006中,鸡蛋备用液需进行两次除脂肪操作,在除脂肪时要求弃去中间由正己烷与水相形成的絮凝层,取全量下层清液过柱净化,由于弃去的絮凝层带走药物,导致回收率偏低。备用液中含有大量的蛋黄成分的脂溶性成分卵磷脂,在除脂肪操作中经激烈的涡旋、振荡容易使正己烷与水相产生乳化,离心后在两相中间形成絮凝层,混合方式越剧烈,形成的絮凝层越厚,备用液损失越多,药物损失也越严重。对比了手动翻转、涡旋、摇床水平振荡等操作,不同的混合方式对下层备用液体积、絮凝层体积和除脂效果有较大影响,具体结果如表6所示。对国标方法农业部 781号公告-6-2006的净化优化方法建议如下:鸡蛋备用液先加入少量纯水定容至6 mL;除脂操作选择中速振荡的方式;为减少检测目标物损失,在第一次除脂中保留中间絮凝层;第二次除脂后只分取3 mL下层备用液进行SPE净化。优化后的方案有效提高了定量结果的准确性,具体回收率对比见图3,其中浅色部分为按照原方法操作并全量过柱得到的回收率,深色部分为按照优化后的净化方法操作得到的回收率。

表6 正己烷与备用液的振摇方式对除脂效果的影响Table 6 Influence of vibration mode of n-hexane and standby liquid on the effect of fat removal

图3 鸡蛋样品回收率对比图Fig.3 Comparison of egg sample recovery
3 结论
本文对农业部1025号公告-14-2008和农业部781号公告-6-2006两个现行有效国标法检测方法的样品前处理提出了优化方案,优化后的方法可以应用于鸡肉、鸡蛋的FQs药物残留检测,步骤简单、可操作性强、准确可靠,能满足生产企业日常自检以及检测机构的批量样品的快速筛查需要。对于需要使用该上述国标方法完成能力验证的检测机构,优化后的方案能为能力验证检测工作多提供一份检测技术参比和质量保障,有助于对检测结果的准确判定。
